










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Gianotti-Crosti (SGC), también llamado acrodermatitis papular de la infancia, es un exantema infrecuente de la infancia que se presenta, por lo general, en niños entre uno y seis años de edad. Este síndrome fue descrito por primera vez en 1955 por Fernando Gianotti, como un exantema asociado al virus de hepatitis B y que se caracteriza por una erupción cutánea, maculopapular, simétrica de distribución acral. 1
En 1970 se estableció la relación de la acrodermatitis papulosa infantil con el virus de la hepatitis B, sin embargo, algunos pacientes presentaron serología frente al virus de la hepatitis B negativa (HBsAg). Por esta razón Gianotti consideró que estos pacientes presentaban un cuadro clínico distinto a la acrodermatitis pustulosa infantil y describieron los síndromes papulovesiculares acro localizados. 2) Algún tiempo después, se encontraron dermatosis con características similares asociadas a otros agentes infecciosos e inmunizaciones, que se nombraron síndromes papulovesiculares acrales y fue en estudios retrospectivos subsecuentes que se demostró, que dichas enfermedades por su clínica son indistinguibles unas de otras y por esta razón en 1979 se decidió agruparlas bajo el nombre de síndrome de Gianotti-Crosti. 3
La fisiopatología del síndrome es desconocida y la causa más frecuente de esta dermatosis se considera viral. 4) La incidencia y prevalencia se desconocen, sin predilección por etnia o género. Sin embargo, dado que los pacientes pueden ser diagnosticados con un exantema viral inespecífico, probablemente es subdiagnosticado. 4
Dado que estos pacientes por lo general son llevados a su pediatra, es importante que el clínico conozca las características fundamentales del síndrome. A continuación se reporta el caso de una paciente con diagnóstico de síndrome de Gianotti-Crosti, también llamado acrodermatitis papular de la infancia.
Presentación del caso
Paciente de 18 meses de edad de piel blanca y sexo femenino, sin antecedentes patológicos personales, ni familiares de interés, que acudió a la consulta porque presentó fiebre de 38 oC que repitió en tres ocasiones, acompañada de un exantema papular localizado en cara, nalgas y las cuatro extremidades. Pasado tres días el exantema se tornó pápulo-vesicular, distribuidos por las cuatro extremidades y glúteos. Como ilustra las (Figuras 1, 2 y 3).

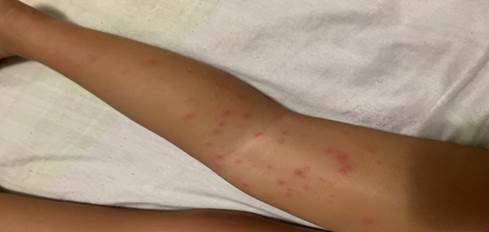
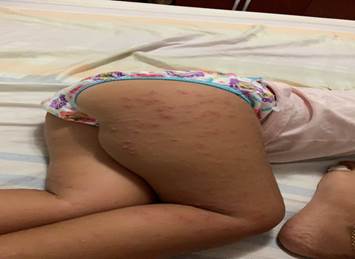
(Imágenes del autor).
El tronco fue respetado y las lesiones no eran pruriginosas. La paciente no presentó otros síntomas, permaneció afebril y sin otra sintomatología. El resto de la exploración física fue normal.
Estudios analíticos:
Leucograma: 12,5 leucocitos x 109/L (60 % de linfocitos).
Velocidad de sedimentación globular (VSG): 13 mm/h.
Transaminasa glutámica pirúvica (TGP): 11 U/l.
Conteo de plaquetas 192 x10-3/L.
Fue diagnosticada de acrodermatitis papulosa. Las lesiones desaparecieron de manera paulatina tras dos semanas de duración, sin tratamiento.
Discusión
El síndrome de Gianotti-Crosti es propio de la infancia de causa viral en la mayoría de los casos, la que se sospechó durante muchos años y en la década de 1970, De Gaspari G et al. 5) confirmaron una asociación con la infección por el virus de la hepatitis B (VHB). Sin embargo, un estudio retrospectivo realizado en 1992 por Caputo R et al. 6) reveló que la asociación con el VHB se encontraba en menos del 25 % de los pacientes. El otro 75 % de los pacientes presentaba otras infecciones virales y no fue posible realizar una distinción clínica entre ambos grupos.
La virosis descrita con mayor frecuencia es la asociada con virus de Epstein-Barr (EBV). Otros virus asociados son citomegalovirus (CMV), coxsackie b, coxsackie A-16, echovirus, hepatitis A, parainfluenza, parvovirus B-19, herpesvirus 6, sincicial respiratorio, paramyxovirus y virus de la parotiditis, 7,8,9,10 además de, vacunas como Bacilo Calmette-Guerin (BCG), antipoliomielítica, antivariolítiva y toxoplasmosis. 11) Existen descripciones en raros eventos de síndrome de Gianotti-Crosti relacionados con bacterias, además de muchos otros sin diagnóstico etiológico. 12,13
Se ha propuesto utilizar el término de síndrome de Gianotti-Crosti, independientemente de cuál sea su causa y forma de presentación para describir todas las dermatosis eruptivas de localización acral. Según su clínica, caracterizadas por lesiones de color rosa o pardo, donde se intercalan lesiones pustulares edematosas o papulovesiculares de 1-10 mm de diámetro que permanecen al menos diez días y que pueden presentar fenómeno de Koebner positivo además de descamación. En este síndrome se observa una dermatitis simétrica distribuida en las mejillas, los glúteos y superficies extensoras de las extremidades, donde el tronco queda libre y rara vez involucra palmas, plantas, áreas flexoras y mucosas. Por lo general, causadas por virus, que siguen un curso benigno y autolimitado de pocas semanas de duración. 14,15
El pediatra, ante cualquiera de las enfermedades que cursan con exantema, está acostumbrado a valorar toda una serie de puntos para llegar a establecer el posible diagnóstico del paciente, con el fin de indicar un pronóstico e implantar las necesidades de su aislamiento, así como aplicar las medidas a adoptar en el entornomás próximo y el posible tratamiento. Estos aspectos son: a) antecedentes personales, epidemiológicos, ingesta de fármacos; b) período de incubación; c) pródromos, síndrome febril, y d) características del exantema, como tipo de lesión, lugar de inicio, distribución posterior, uniformidad o diversidad de las lesiones, momento evolutivo de las mismas y clínica acompañante. 16
La acrodermatitis papular infantil se debe entender como la presencia de lesiones cutáneas monomorfas, de tipo papular o papulovesicular, con superficie plana, de color entre rosado y marrón, con un diámetro de 1-10 mm, de número muy variable, con disposición simétrica en las mejillas, las nalgas, las superficies de extensión de las extremidades superiores e inferiores y con una duración entre tres y cinco semanas. Si bien esta sería la forma típica de presentación, existen pequeñas variaciones que pueden acompañar a algunos casos: a) la posibilidad de confluencia de lesiones; b) prurito que suele acompañar a las lesiones papulovesiculares, y c) la localizaciónen pabellones auriculares, presente en los primeros días de las manifestaciones y que suele asociarse también con las formas papulovesiculares.
Los aspectos clínicos negativos que ayudan al diagnóstico son la inexistencia de las lesiones en el tronco y la presentación de lesiones descamativas. No puede obviarse la presentación de un episodio febril de poca intensidad, unos 7-10 días antes, acompañado de manifestaciones catarrales de las vías respiratorias altas. 17
El diagnóstico es clínico y se apoya en el antecedente de infección de las vías aéreas superiores, además de síntomas generales como mal estado en general, fiebre de bajo grado, linfadenopatía cervical, axilar o inguinal, hepatomegalia o esplenomegalia. Por laboratorio se puede presentar linfopenia o incluso linfocitosis moderada, además de monocitosis en los casos relacionados con EBV y transaminasas elevadas asociadas a EBV y CMV. 4
La enfermedad se encuentra infradiagnosticada dado que es fácil confundirla con otras lesiones pápulo vesiculares en la infancia. El diagnóstico diferencial se debe establecer con los siguientes procesos: acrodermatitis enteropática, eritema infeccioso, eritema multiforme, enfermedad mano-pie-boca, púrpura de Schönlein-Henoch, enfermedad de Kawasaki, liquen plano, síndrome papular purpúrico en guante y calcetín, urticaria papular, escabiosis, enfermedad de Letterer-Siwe y reacciones cutáneas a medicamentos. Otros diagnósticos diferenciales incluyen prurigo agudo, granuloma anular, dermatitis atópica, pitiriasis liquenoide varioliforme aguda y pitiriasis rosada. 18
Las alteraciones histopatológicas pueden ser dramáticas pero no son específicas. Se dividen en alteracionesde patrón vesicular y patrón no vesicular; en la primera se observa epidermis con acantosis moderada acompañada de espongiosis difusa y vesículas, donde las células predominantes son de Langerhans y en ocasiones se observa exocitosis linfocitaria, imitando una micosis fungoide; en la dermis papilar se ve un infiltrado linfocitario perivascular intenso con predominio de linfocitos T y células dentríticas. En la forma no vesicular, la epidermis puede tener leve acantosis y paraqueratosis focal con espongiosis, en la dermis se observa una superficie perivascular irregular e incluye paraqueratosis focal, edema en la dermis papilar, inflamación perivascular superficial con infiltración linfocitaria con algunos histiocitos y en ocasiones dermatitis sobre la capa superficial, dermatitis liquenoide o vasculitis linfocítica con hemorragia. 4
La mayoría de los pacientes tienen un curso benigno y autolimitado con desaparición de las lesiones cutáneas entre 10 y 60 días posteriores al inicio del cuadro, por lo general resuelve sin secuelas y rara vez con hipopigmentación o hiperpigmentación postinflamatoria. La hepatoesplenomegalia puede desaparecer al mismo tiempo que las lesiones cutáneas, pero las adenopatías se pueden encontrar varios meses después. 14
Debido a la evolución benigna del síndrome de Gianotti-Crosti y a su curso autolimitado con poca sintomatología, no es necesario un tratamiento específico en los pacientes, por lo que solo se puede limitar a tratamiento sintomático, sobre todo en los casos en que las lesiones produzcan prurito el tratamiento puede ser a base de antihistamínicos orales. 17
Conclusiones
El síndrome de Gianotti-Crosti es un exantema infeccioso que se presenta sobre todo en niños de entre uno y seis años de edad, rara vez se observa en adultos. La presentación clásica es la de un exantema simétrico de distribución acral, que por tratarse de un exantema viral inespecífico se suele subdiagnosticar. En general la dermatosis suele ser asintomática o enalgunos casos presenta prurito leve, como en el caso que se presentó. La evolución tiene un curso benigno y autolimitado, con resolución de las lesiones hacia la tercera semana

