










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkVaccimonitor
versión impresa ISSN 1025-028X
Vaccimonitor vol.22 no.1 Ciudad de la Habana ene.-abr. 2013
ARTÍCULO ORIGINAL
Seguridad del Racotumomab en el tratamiento de pacientes con cáncer de pulmón de células no pequeñas
Safety of Racotumomab in the treatment of patients with non-small cell lung cancer
Leslie Pérez,* Daymys Estévez, Yoisbel Gastón, Amparo Macías, Carmen Elena Viada
Centro de Inmunología Molecular (CIM). Calle 216, esq 15. Atabey, Playa, La Habana, Cuba.
email:leslie@cim.sld.cu
* Lic. Farmacia, MSc, Investigador agregado.
RESUMEN
En Cuba, el cáncer de pulmón es el segundo en incidencia y el primero en mortalidad. Por tanto, se necesita identificar nuevas opciones terapéuticas. Los enfoques inmunológicos son interesantes debido al potencial de actividad sin las toxicidades de la quimioterapia convencional. El Centro de Inmunología Molecular generó una vacuna denominada Racotumomab, la cual actúa sobre el carcinoma pulmonar, aumentando la apoptosis tumoral y disminuyendo la cantidad de vasos tumorales. Para evaluar su seguridad se realizó un estudio de acceso expandido, multicéntrico y abierto en 86 pacientes con cáncer de pulmón de células no pequeñas. La dosis administrada fue de 1 mg/mL por vía intradérmica. Las cinco primeras dosis se administraron cada 14 días y las restantes 10 cada 28 días, hasta completar el año de tratamiento. Las reinmunizaciones durante el seguimiento fueron cada 28 días. Se analizó la aparición de los eventos adversos y se clasificaron acorde con los criterios de la CTC v4.02. Estos se reportaron en 58 pacientes (67,4%), para un total de 215 eventos; el más frecuente fue el ardor en el sitio de la inyección, 32 (14,9%). El uso de la vacuna en los pacientes estudiados evidenció buen nivel de seguridad y tolerancia.
Palabras clave: Racotumomab, ensayo clínico, cáncer de pulmón de células no pequeñas, eventos adversos.
ABSTRACT
In Cuba, lung cancer ranks second in incidence and first in mortality. Therefore, it is necessary to identify new therapeutical options. Immunological approaches are interesting because of the potential activity without the toxicities of conventional chemotherapy. The Center of Molecular Immunology developed a vaccine called Racotumomab; it acts on the lung carcinoma inducing an increase in tumor apoptosis and a decrease in the number of tumor vessels. A expanded access, multicenter, open study was conducted in 86 patients with non-small cell lung cancer in order to assess its safety. The administered dose was 1 mg/mL intradermically. The first 5 doses were administered every 14 days and the remaining 10 every 28 days until completing the treatment. The follow-up re immunizations were every 28 days. The occurrence of adverse events (AE) was analyzed and they were classified according to CTC v4.02 criteria. Adverse events were reported by 58 patients (67.4%), making a total of 215 events. burning at the injection site was the most frequently reported event, 32 (14.9%). The use of the vaccine in the patients under study showed good safety and tolerance.
Key words: Racotumomab, clinical trial, non-small cell lung cancer, adverse events.
INTRODUCCIÓN
A escala mundial el cáncer de pulmón es la neoplasia maligna más frecuente, con un estimado de 1,35 millones de casos nuevos cada año; también es la localización de cáncer que más muertes causa con 1,18 millones de muertes anuales, para una razón de mortalidad/incidencia de 0,87 (1).
En el 2008 se estimó que aproximadamente 215.000 nuevos casos de cáncer de pulmón serían diagnosticados en Estados Unidos (2). El tipo histológico más frecuente es el cáncer de pulmón de células no pequeñas (CPCNP), que constituye aproximadamente el 80% de todos los casos nuevos. El mismo incluye tres subtipos histológicos: epidermoide, adenocarcinoma y carcinoma de células grandes.
En Cuba, dentro de las neoplasias, el cáncer del pulmón es el primero en mortalidad y el segundo en incidencia. Según el Anuario Estadístico 2010, del Ministerio de Salud Pública (MINSAP), actualizado con datos del Registro Nacional de Cáncer, la tasa de incidencia de cáncer de pulmón en el año 2007 en el sexo masculino fue de 60,3/100.000 habitantes, mientras que en el sexo femenino fue de 30,5/100.000 habitantes, produciéndose en el año 2010 unas 5.100 defunciones como consecuencia de esta neoplasia (3.274 en el sexo masculino y 1.826 en el femenino), con una tasa de 45,4% (3).
El Centro de Inmunología Molecular (CIM), de La Habana, desarrolló una vacuna que contiene un anticuerpo antiidiotipo monoclonal murino, perteneciente a la subclase IgG1, denominado Racotumomab (1E10/hidróxido de alúmina). Este se obtiene inmunizando ratones Balb/c con el AcM P3, que reconoce los gangliósidos que tienen ácido siálico N-glicolilado, glucolípidos sulfatados y antígenos expresados en melanomas y carcinomas de mama y pulmón (4-6).
El Racotumomab muestra un efecto antitumoral, tanto en animales de experimentación (8) como en los ensayos clínicos efectuados en diferentes localizaciones, incluida el cáncer de pulmón de células no pequeñas (5, 8, 9).
Teniendo en cuenta que esta vacuna es un producto en investigación, se llevó a cabo este estudio en los pacientes diagnosticados con cáncer de pulmón de células no pequeñas, incluidos en el protocolo de acceso expandido llevado a cabo por el CIM, con el objetivo evaluar su seguridad y tolerancia.
MATERIALES Y MÉTODOS
Se evaluaron los eventos adversos presentados en 86 pacientes que recibieron tratamiento con Racotumomab durante el desarrollo del protocolo de acceso expandido, multicéntrico y abierto.
Los sujetos fueron reclutados en la consulta de Oncología de los hospitales cubanos: "Celestino Hernández Robau", "Hermanos Ameijeiras", "José Ramón López Tabranes" y "Saturnino Lora", con diagnóstico citohistológico de CPCNP, que al momento de la inclusión en el estudio presentaban progresión de la enfermedad después de la primera línea estándar de tratamiento oncoespecífico.
La vacuna Racotumomab (dosis de 1 mg/mL) se administró por vía intradérmica con una jeringa descartable de 1 mL y agujas hipodérmicas atóxicas, libres de pirógenos, de calibre 25-27G".
Cada dosis individual se dividió en cuatro subdosis intradérmicas (de 0,25 mL), administradas cada una de ellas en un lugar diferente, seleccionado a criterio del investigador: entre la región deltoidea, la cara anterior del antebrazo, la cara anterior del muslo y la cara posterior de la pantorrilla. Los pacientes recibieron 15 inmunizaciones como tratamiento de base, las primeras cinco cada 14 días (fase de inducción) y las restantes 10 cada 28 días (fase de mantenimiento).
Las reinmunizaciones durante el período de seguimiento, después de terminado su tratamiento de base, fueron administradas cada 28 días. Se continuó la vacunación mientras los pacientes tuvieran un estado general favorable y no toxicidad inaceptable.
Para la evaluación de la toxicidad se recopilaron todas las evidencias de eventos adversos reportados durante el estudio a través del interrogatorio y el examen físico realizado a los pacientes, monitoreo de los signos vitales con cada administración de los diferentes tratamientos y mediante los exámenes de laboratorio clínico.
La clasificación de la intensidad de los eventos adversos se realizó utilizando los criterios comunes de toxicidad (CTC, del inglés Common Toxicity Criteria) versión 4.02 planteados por el Instituto Nacional de Cáncer de los Estados Unidos. La evaluación de la relación de causalidad entre el evento adverso y el tratamiento en investigación contempló las categorías siguientes:
A. No relacionado: Existe suficiente información para establecer que la etiología del evento no está relacionada con el medicamento.
B. Posible: Puede ser un evento adverso con una secuencia temporal a la administración del medicamento, que sigue un patrón conocido y esperado, de acuerdo con la experiencia preclínica y clínica con dicho medicamento y similares, pero que no mejoró al interrumpir la administración o reducir la dosis; o que mejoró al interrumpir la administración o reducir la dosis, pero que se explica por el estado clínico del sujeto.
C. Probable: Puede ser un evento adverso con una secuencia temporal a la administración del medicamento, que sigue un patrón conocido y esperado, de acuerdo con la experiencia preclínica y clínica con dicho medicamento y similares, que no pudo ser explicado por el estado clínico del sujeto y que es confirmado por una mejoría al interrumpir la administración o reducir la dosis.
D. Muy Probable: Puede ser un evento adverso con una secuencia temporal a la administración del medicamento, que sigue un patrón conocido y esperado de acuerdo con la experiencia preclínica y clínica con dicho medicamento y similares, que es confirmado por una mejoría al interrumpir la administración o reducir la dosis, que reaparece al reiniciar la terapia, pero pudo ser explicado por el estado clínico del sujeto.
E. Definitiva: Puede ser un evento adverso de una secuencia temporal a la administración del medicamento, que sigue un patrón conocido y esperado de acuerdo con la experiencia preclínica y clínica con dicho medicamento y similares, que no pudo ser explicado por el estado clínico del sujeto, que es confirmado por una mejoría al interrumpir la administración o reducir la dosis y que reaparece al reiniciar la terapia.
Las categorías A y B se consideran no relacionadas con el producto; las C, D y E se consideran relacionadas (reacciones adversas del producto).
Para clasificar un evento adverso como serio se tuvo en cuenta el resultado de este para el paciente o los criterios de actuación asociados a los tipos de eventos que constituyen una amenaza para la vida del paciente o para su funcionamiento, considerándose como evento adverso serio cualquier suceso desfavorable que resultara en fallecimiento, amenazara la vida, demandara la hospitalización del paciente o la prolongara, diera como resultado incapacidad/invalidez persistente o significativa, o trajera como consecuencia una anomalía congénita/defecto de nacimiento.
En el análisis estadístico de la seguridad se incluyeron los sujetos que recibieron al menos una dosis de medicamento y se determinó la frecuencia de pacientes que presentaron eventos adversos. Se hicieron tablas de contingencia de dos entradas relacionando el tipo de reacción con la intensidad del evento, relación de causalidad, duración del evento, actitud respecto al fármaco y tratamiento aplicado.
El protocolo se diseñó y ejecutó según los principios éticos para la investigación médica con sujetos humanos, establecidos en la actualización de la Declaración de Helsinki durante la Asamblea General de la Asociación Médica Mundial, efectuada en Seúl, Corea del Sur, octubre de 2008. También se tuvieron en cuenta las regulaciones estatales vigentes en la República de Cuba para la ejecución de este tipo de investigación.
Por ser una investigación con seres humanos, el protocolo del ensayo fue revisado previamente y aprobado para su ejecución desde el punto de vista metodológico, científico y ético por los Comités de Ética institucionales de los hospitales participantes.
Al ser un estudio de acceso expandido no requirió la aprobación del Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED), sino la comunicación de inicio del mismo a la agencia.
RESULTADOS
La etapa de inclusión tuvo una duración de 20 meses, en la cual se incluyeron 86 pacientes tratados con la vacuna Racotumomab. Todos los pacientes cumplieron con los criterios de selección establecidos en el protocolo.
Se incluyeron los pacientes reclutados por cuatro instituciones hospitalarias de cuatro provincias de Cuba, estableciéndose el ritmo de inclusión para cada una, previo inicio del estudio, por referencia de los investigadores con relación a la incidencia de pacientes con la patología en estudio. El mayor número de incluidos fue reportado en el hospital "Celestino Hernández", con un 61,62% del total, seguido del "José Ramón López Tabranes", con un 25,6%.
La muestra de pacientes estuvo caracterizada por predominio de color de piel blanco con 64 (74,42%), promedio de edad de 62 años, sexo masculino 65 pacientes (75,6%). De acuerdo con la clasificación estipulada por el Grupo Oncológico Cooperativo del Este de los Estados Unidos, existió un predominio de ECOG 0-1, con 73 pacientes (84,9%).
Con respecto al tipo histológico, prevaleció el carcinoma epidermoide en 42 pacientes (48,8%). Referente al estadio de la enfermedad, la mayoría de los pacientes fueron clasificados al momento del diagnóstico como avanzados (estadios IIIb 43, 50% / IV 31, 36,05%), lo cual coincide con lo reportado en el estado del arte para esta indicación clínica. Como tratamiento previo, la mayoría de los pacientes recibieron quimioterapia, 72 (83,7%).
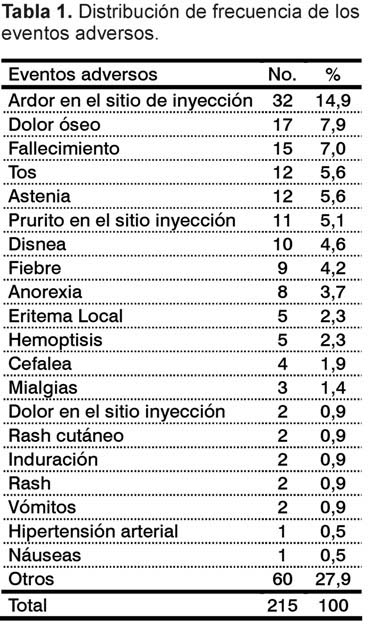
De los 86 pacientes que recibieron el preparado vacunal, 58 (67,4%) reportaron eventos adversos relacionados con las administraciones, para un total de 215 eventos.
En nuestra muestra el evento adverso más frecuente fue el ardor en el sitio de inyección; otros eventos adversos con menor frecuencia de aparición fueron: dolor óseo (se produjo desde la primera dosis y con dosis posteriores, generalmente duró entre 1 y 3 días, con una recuperación completa), tos, astenia, prurito en el sitio de inyección, disnea (de aproximadamente 2 h de duración en la mayoría de los pacientes) y fiebre. Los restantes eventos adversos englobados en la categoría "otros" tuvieron una frecuencia de presentación menor al 2% (Tabla 1). La mayoría de las reacciones locales desaparecieron de 4 a 7 días.
La clasificación de intensidad de todos los eventos adversos se hizo acorde con lo establecido en la CTC, versión 4,02. El mayor porcentaje estuvo en la categoría leve 150 (69,76%); en menor cuantía las categorías de moderada 23 (10,69%); severa 16 (7,44%) y muy severa 26 (12,09%).
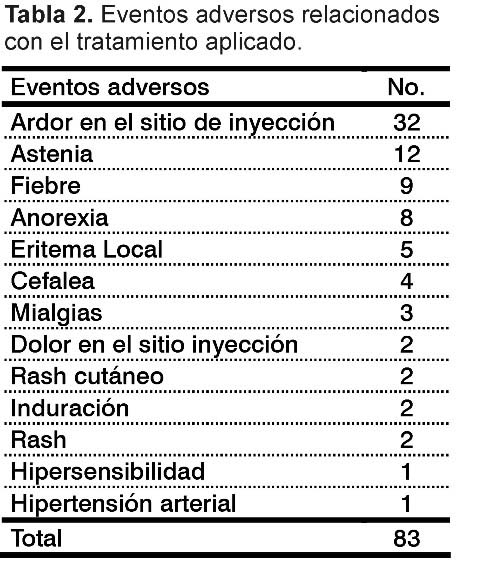
Referente a la relación de causalidad, se clasificaron como definitiva 34 eventos adversos (16,03%), 40 como probable (18,86%), 8 muy probable (3,77%) y 1 como posible (0,471%), para un total de 83 eventos adversos relacionados con el tratamiento (39,15%) (Tabla 2).
Como no relacionados fueron clasificados 132 eventos (61,39%).
Con respecto al evento adverso hipertensión arterial, la misma ocurrió después de administrado el producto y fue controlada con captopril (25 mg). El evento adverso muerte no estuvo relacionado con la administración del producto y sí con el estado clínico de los pacientes.
En cuanto a la intensidad de los eventos adversos clasificados como relacionados con el tratamiento, el 100% (83 eventos) fue catalogado como ligero, lo que corresponde a los grados I-II, según la escala de la clasificación utilizada. La mayoría de los eventos fueron de corta duración (2 a 3 días) y no requirieron interrupción del tratamiento.
DISCUSIÓN
La clasificación de los eventos adversos se realiza utilizando los criterios comunes de toxicidad del Instituto Nacional de Cáncer de los Estados Unidos (10). Esta es la escala de toxicidad más empleada en los ensayos clínicos de Oncología.
Los eventos adversos reportados con mayor frecuencia y relacionados con el preparado vacunal se refieren, fundamentalmente, a los observados en el área o sitio de la administración y a los llamados síntomas gripales que habitualmente se producen posterior a las administraciones inmunoterapéuticas (11, 12).
El dolor es el evento adverso más frecuentemente reportado de conjunto con ardor en el sitio de inyección, eritema local, disnea, astenia, induración, cefalea, dolor óseo, mialgia, artralgia, anorexia, vómitos, náuseas, fiebre, tos, prurito, dolor abdominal e hipertensión arterial (9). Lo que concuerda con los resultados de este estudio.
El ardor en el sitio de inyección está relacionado con la forma de administración propia del medicamento y con las características del adyuvante utilizado. El mismo es un ardor ligero, por lo que es tolerable y no limita que el paciente pueda continuar con el esquema de tratamiento.
La hemoptisis en este estudio no se encuentra reportada en la literatura consultada. El mismo fue clasificado como no relacionado con el producto vacunal, así como de intensidad ligera, por lo que fue tolerable y no impidió que los pacientes pudieran continuar y cumplimentaran el esquema de tratamiento previsto.
En un estudio fase I realizado en Argentina en pacientes con cáncer de mama, una paciente tratada con 1 mg desarrolló una mácula dolorosa con una vesícula seguida de ulceración, luego de recibir la quinta dosis. Una biopsia de piel reveló vasculitis y la paciente fue retirada de la terapia. Otra paciente sufrió un síncope 10 días después de recibir la novena dosis de 2 mg. Este evento se consideró de improbable relación con la vacuna, pero la paciente interrumpió el tratamiento (16). Estos eventos no se presentaron en nuestra población estudiada.
La presencia de pápulas en el sitio de inoculación se observó en estudios preclínicos con ratas Cenp: SPRD de ambos sexos, procedentes de la División de Roedores Gonotobióticos del Centro Nacional para la Producción de Animales de Laboratorio (CENPALAB), tratadas con 0,1 mL de vacuna antiidiotípica o 0,1 mL de gel de alúmina (grupo vehículo). Lo cual nos indica que posiblemente este evento esté relacionado con el gel de alúmina que también está presente como adyuvante de la vacuna (20).
En el estudio preclínico de tolerancia local realizado con ratas albinas de la línea Wistar, provenientes del CENPALAB, de acuerdo con los estándares planteados por la Comunidad Europea, tratadas con 0,1 mL de la vacuna o 0,1 mL de gel de alúmina, en el sitio de administración se detectó la presencia de granulomas. Desde el punto de vista macroscópico no se observaron reacciones edematosas, eritematosas o de formación de escaras en el sitio de administración o regiones circundantes (20).
De manera general la vacuna antiidiotípica fue bien tolerada, tal y como lo describen otros estudios (20). Los eventos adversos relacionados con el producto en investigación se resumen, fundamentalmente, a los observados en el sitio de aplicación y a los llamados síntomas generales que habitualmente se producen con este tipo de terapia.
Los eventos adversos, en general, no conllevaron a la interrupción de las administraciones ni tuvieron repercusión grave sobre la salud de los pacientes y según los síntomas fueron controlados satisfactoriamente con fármacos, tales como: acetaminofen, ibuprofeno, difenhidramina, dipirona, captopril, entre otros.
Hasta el momento no se han reportado granulomas, nódulos ni descamación en el sitio de la inyección.
Este estudio coincide con lo observado en ensayos anteriores, en los cuales los eventos adversos más frecuentes relacionados con el uso de la vacuna Racotumomab son locales, cuya intensidad es leve y moderada. No se detectaron eventos adversos graves relacionados con la utilización del preparado vacunal. Por lo que se concluye que el tratamiento con la misma es seguro.
REFERENCIAS
1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74-108.
2. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96.
3. Ministerio de Salud Pública, Dirección Nacional de Registros Médicos y Estadísticas de Salud. Anuario Estadístico de Salud 2010. La Habana: MINSAP; 2011.
4. Neninger E, Díaz R, de la Torre A, Rives R, Díaz A, Suárez G, et al. Active immunotherapy with 1E10 MAb anti-idiotype vaccine in patients with small cell lung cancer. Report of a phase I trial. Cancer Biol Ther 2007;6:145-50.
5. Alfonso M, Díaz A, Hernández AM, Pérez A, Rodríguez E, Bitton R, et al. An antiIdiotype vaccine elicits a specific response to N-Glycolyl sialic acid residues of glycoconjugates in melanoma patients. J Immunol 2002;168:2523-9.
6. Hernández AM, Rodríguez M, López-Requena A, Beausoleil I, Pérez R, Vázquez AM, et al. Generation of anti-Neu-glycolyl-ganglioside antibodies by immunization with an anti-idiotype monoclonal antibody: A self-versus non-self-matter. Immunobiology 2005;210(1):11-21.
7. Vázquez AM, Gabri M, Hernández AM, Alonso DF, Beausoleil I, Gómez DE, et al. Antitumor properties of an anti-idiotypic monoclonal antibody in relation to N-glycolyl-containing gangliosides. Oncol Rep 2000;7:751-6.
8. Díaz A, Alfonso M, Alonso R, Suárez G, Troche M, Catalá M. Immune responses in breast cancer patients immunized with an anti-idiotype antibody mimicking NeuGc-containing gangliosides. Clin Immunol 2003;107(2):80-9.
9. Alfonso S, Díaz RM, de la Torre A, Santiesteban E, Aguirre F, Pérez K, et al. 1E10 Anti-idiotype Vaccine in Non-Small Cell Lung Cancer. Experience in Stage IIIb/IV Patients. Cancer Biol Ther 2007;6:1847-52.
10. U.S Department of Health and Human Services, National Institutes of Health, National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE). Version 4.0. Washington: National Institutes of Health-National Cancer Institute; 2009.
11. Schiller JH, Harrington D, Belani CP, Sandler A, Langer C, Sandler A, et al. A randomized phase III trial of four chemotherapy regimens in advanced non-small cell lung cancer. N Engl J Med 2002;346:92-8.
12. Carr A, Rodríguez E, Arango MC, Camacho R, Osorio M, Gabri M, et al, Immunotherapy of Advanced Breast Cancer With a Heterophilic Ganglioside (NeuGcGM3) Cancer Vaccine. J Clin Oncol 2003;21:1015-21.
13. Toledo D, Alfonso S, Santiesteban E, Aguirre F, Hernández AM, Mazorra Z, et al. La Vacuna Anti-idiotipo 1E10: Experiencias en la Inmunoterapia del Cáncer Avanzado. Cancerología 2009:4(3):143-6.
Recibido: Julio de 2012
Aceptado: Septiembre de 2012

{kind=link}
{kind=link}