










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.33 no.3 La Habana jul.-set. 2016
RESEARCH
The role of promoter DNA methylation of six cancer-associated miRNA genes in ovarian cancer development and progression
Rol de la metilación en el ADN del promotor de seis genes de miARN asociados al cáncer en el desarrollo y progresión del cáncer de ovario
Vitaly I Loginov1, Alexey M Burdennyy1, Irina V Pronina1,2, Elena A Filippova1, Tatiana P Kazubskaya3, Eleonora A Braga1
1 Laboratory of Pathogenomics and Transcriptomics, Institute of General Pathology and Pathophysiology, Moscow, 125315, Russia.
2 Laboratory of Molecular Genetics of Complex Inherited Diseases, Research Center of Medical Genetics, Moscow, 115478, Russia.
3 Laboratory of Clinical Oncogenetics, Research Institute of Clinical Oncology, N.N. Blokhin Russian Cancer Research Center, Moscow, 115478, Russia.
ABSTRACT
DNA methylation of promoter CpG islands and interactions between microRNAs and messenger RNAs of target genes are considered two crucial mechanisms for gene and pathway deregulation in malignant tumors. Methylation was analyzed for the large group of tumor-suppressor microRNA genes in various malignancies, but in ovarian tumors, it was not studied extensively yet. Our goal was to identify novel microRNA genes with aberrant methylation in epithelial ovarian cancer and assess contribution of promoter methylation of these genes to ovarian cancer development and progression. Therefore, DNA methylation of six microRNA genes (MIR-9-1, MIR-9-3, MIR-132, MIR-148a, MIR-191, MIR-212) was analyzed using a representative set of 54 paired (tumor/normal) ovarian tissue samples and methylation-specific PCR. The methylation frequencies of four of the examined genes (MIR-9-1, MIR-9-3, MIR-132, MIR-148a) were significantly higher in tumor samples in comparison with matched histologically normal samples: 20-57 % vs 4-9 % (p ≤ 0.01, Fisher’s exact test). Conversely, hypomethylation was revealed for MIR-191: 13 % vs 59 %, p ≤ 0.01. We showed strong association of MIR-9-1 and MIR-9-3 hypermethylation with advanced III/IV clinical stages, low differentiation, high tumor sizes (p ≤ 0.05), and the presence of lymph node metastases on the level of a trend. Besides, we observed the dependence of MIR-191 hypomethylation on the ovarian tumor cell differentiation (p ≤ 0.05). Thus, our findings provide the evidence on the considerable contribution of aberrant methylation of five microRNA genes to ovarian cancer development and progression, and allow us to suggest novel potential biomarkers.
Keywords: DNA methylation, miRNA genes, epithelial ovarian cancer, cancer progression, microRNA.
RESUMEN
Aunque la metilación del ADN en las islas de CpG de los promotores y su interacción con los micro ARN (miARN) de sus genes blanco son mecanismos cruciales para la desregulación de genes y rutas genéticas en tumores malignos, su ocurrencia y efecto han sido estudiados escasamente en genes de miARN supresores en tumores de ovario. El propósito de este estudio fue identificar nuevos genes de miARN con metilación aberrante en cáncer epitelial de ovario y evaluar su efecto sobre el desarrollo y progresión del cáncer. Para ello se estudió la metilación en el ADN de seis miARN (MIR-9-1, MIR-9-3, MIR-132, MIR-148a, MIR-191, MIR-212) en muestras pareadas de tejido (tumoral/normal) de ovario y mediante PCR específico para la metilación. Las frecuencias de metilación en los cuatro primeros genes fueron significativamente altas en las muestras tumorales comparadas con las del tejido normal equivalente: 20-57 % vs 4-9 % (p ≤ 0.01, prueba exacta de Fisher). Por el contrario, se detectó hipometilación para el MIR-191: 13 % vs 59 %, p ≤ 0.01. Hubo una alta asociación de la hipermetilación en MIR-9-1 y MIR-9-3 con estadios clínicos avanzados III/IV, baja diferenciación, alta talla tumoral (p ≤ 0.05), y una tendencia en la metástasis hacia nódulos linfáticos. La diferenciación de células tumorales de ovario fue dependiente de la hipometilación de MIR-191 (p ≤ 0.05). La metilación aberrante en cinco genes de miARN contribuyó sustancialmente al desarrollo y la progresión del cáncer de ovario, y permite sugerir su uso como un nuevo y potencial sistema de biomarcadores.
Palabras clave: metilación de ADN, genes miARN, cáncer epitelial de ovario, progresión del cáncer, miARN.
INTRODUCTION
Epithelial ovarian cancer (EOC) is the most lethal of malignant gynecological tumors and the fifth leading cause of death resulting from cancer among women [1]. The major reason for this high mortality rate is the lack of early detection methods. Indeed, the five-year survival rate in cases of stage I-II EOC is estimated to be approximately 90 % [2]. These data demonstrate the relevance of investigations on gene expression regulation in EOC pathogenesis, which can lead to the identification of novel molecular markers and targets for therapy of EOC [3], hypermethylation among them.
DNA methylation is one of the most important epigenetic modifications of the genome involved in the regulation of numerous cellular processes through gene silencing without altering DNA sequences. The selective hypermethylation of CpG islands of promoter regions of protein-coding genes, which possess the properties of a tumor-suppressor, is observed in cancer [4]. MicroRNAs (miRNAs), a class of single-stranded noncoding RNAs of 19-25 nucleotides in length, function as post-transcriptional regulators of expression of corresponding target protein-coding genes. The methylation of CpG islands overlapping promoter regions is also involved in the regulation of miRNA gene expression. Of interest, miRNA gene methylation was observed five to ten times more frequently than that of protein-coding genes [5]. Methylation may reduce the ability of miRNAs to inhibit target genes by down-regulation miRNA gene expression and, thus, significantly affect the regulation of signaling pathways and processes involved in tumorigenesis.
Recently, dysregulation of tumor-suppressor miRNAs mediated by promoter DNA hypermethylation was shown to be implicated in major blood and solid human cancers, including melanoma, acute myeloid leukemia, chronic lymphocytic leukemia, colorectal, gastric, lung, breast, bladder and other cancers [6, 7]. Moreover, it appears that methylated miRNA genes could be potential biomarkers for diagnosis or therapy of various malignancies. Now, epigenetic therapies, which goal is to reverse the changes, are standard in case of preleukemic disorder and form of lymphoma, but application of epigenetic therapies in treatment of solid tumors is also emerging as a viable therapeutic route [8]. Epigenetic markers have not been studied widely in such tumors as EOC. Data on hypermethylation of miRNA genes in EOC are just in a few reports so far [9-12].
Therefore, this work was aimed to analyze DNA methylation of six cancer-associated miRNA genes (MIR-9-1, MIR-9-3, members of the MIR-132/-212 cluster, MIR-148a, and MIR-191), which play an important role in the tumorigenesis of epithelial tumors, using a representative set of EOC samples. At the time of writing, there was just a single work concerning the methylation of these genes in ovarian cancer, which reported the involvement of MIR-9-1 and MIR-9-3 hypermethylation in EOC progression and drug resistance of patients [10].
MATERIALS AND METHODS
Tissue and DNA samples
In total, 54 paired tumor/normal samples of EOС were obtained from the N.N. Blokhin Cancer Research Center (Moscow, Russia) in the period. The samples were collected in accordance with the guidelines issued by the Ethics Committee of the N.N. Blokhin Cancer Research Center. All patients gave written informed consent (available upon request). The study was performed in accordance with the principles outlined in the Declaration of Helsinki [13]. Tumor tissues and matched histologically normal tissues were obtained from patients after surgical resection and prior to radiation or chemotherapy, and they were stored in liquid nitrogen. Diagnoses were verified by histopathology, and only samples containing 70-80 % or more tumor cells were used in the studies. ‘Normal’ controls were also confirmed histologically to be normal epithelial cells. Tumor samples from 54 women were characterized according to the International System of Classification of Tumors based on the tumor-node-metastasis (TNM) and staging classification of the Union for International Cancer Control (UICC, version 2002) [14] and the World Health Organization (WHO) criteria classification [15]. Nitrogen-frozen tissues were disrupted using a Mikro-Dismembrator (Sartorius, Germany). The DNA from human tissues was isolated using phenol extraction according to standard protocols.
Methylation specific PCR (MSP)
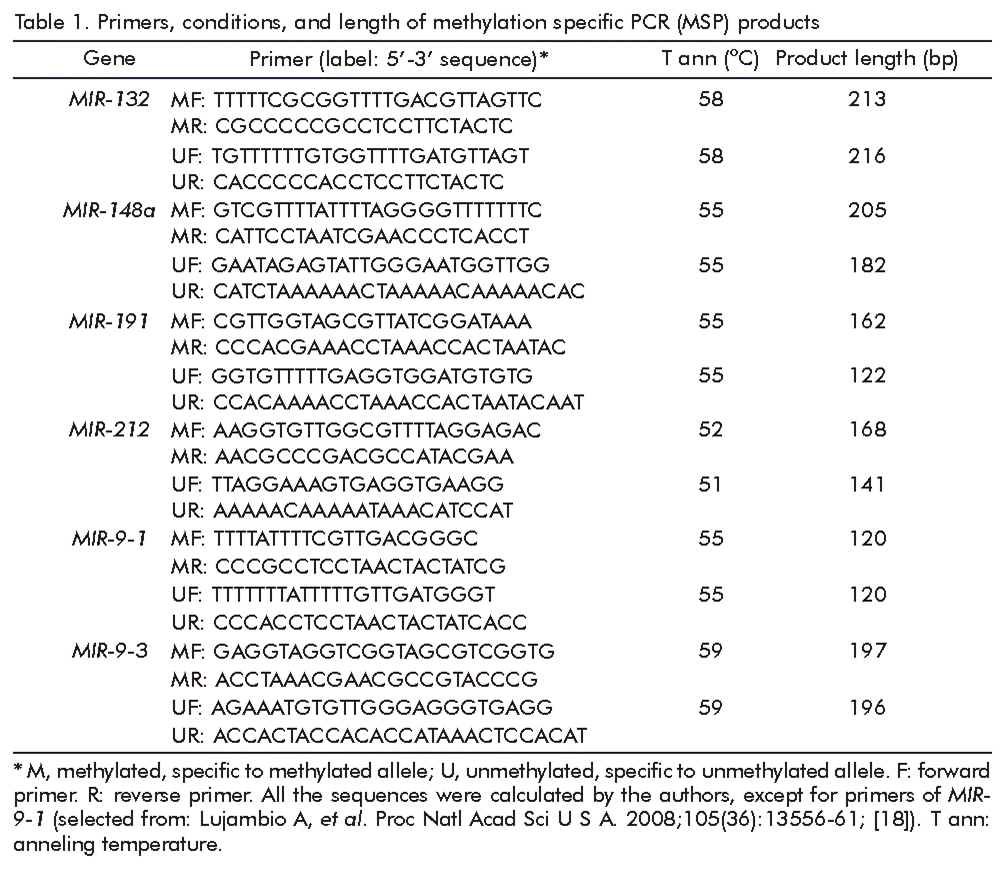
Bisulfite DNA conversion was performed as described [16, 17] using 1-2 μg of DNA. The modified DNA was purified using a Centrifugal Filter Microcon Unit and Ultracel Discs YM-30 (Millipore, USA). Modified DNA was maintained at -20 °C and used as a template for PCR with the designed primers (Table 1). Primers were custom design, except for the MIR-9-1 [18]. From three to six CpG sites were analyzed for each gene.
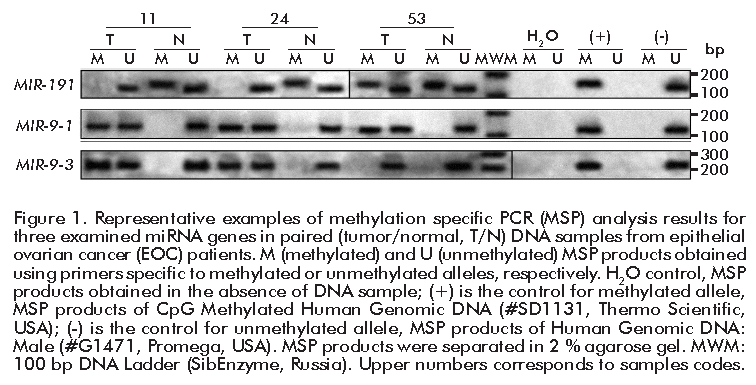
PCR was performed using a DNA Engine Dyad Cycler amplifier (Bio-Rad, United States) with the following program: one cycle at 95 °C for 5 min; 35 cycles at 95 °C for 10 s, annealing temperature (Table 1) for 20 s and 72 °C for 30 s and one cycle at 72 °C for 3 min. CpG Methylated Human Genomic DNA (#SD1131, Thermo Scientific, USA) served as control for methylated allele, while DNA from the Human Genomic DNA: Male (#G1471, Promega, USA) as control for unmethylated allele. False positive results of incomplete bisulfite treatment were excluded on the stage of primer selection if there was no PCR product with untreated DNA and MSP primers (both for methylated and unmethylated alleles) (Figure 1).
PCR products from gene fragments were simultaneously separated on 2 % agarose gels. To determine the intensity of PCR product luminescence, the Gel DOC Ez Imager software (BioRad, USA) was used. Samples with signals approximately equivalent to those of the marker (7 ng/μL) were scored as methylated. Samples with faintly positive signals were repeated three times, and only samples with consistently positive signals were scored as methylated. To establish the sensitivity of the MSP, 1 μg of methylated control DNA was 10-fold serially diluted in buffer, bisulfite-treated and amplified with MSP primers. Hypermethylation was determined to be present in case when methylation was detected in the tumor sample but no methylation was detected in the matched normal sample. Hypomethylation was determined to be present in the inverse case. Retention was determined to be present when no methylation was detected in both tumor and matched normal samples or methylation was detected in both tumor and matched normal samples. Cases with methylation in both tumor and matched normal samples were assumed to be rare.
Statistical analysis
Data was statistically analyzed by using the Fisher’s exact test, with differences considered significant at p ≤ 0.05.
RESULTS
Methylation profiles of 6 cancer-associated miRNAs in EOC
DNA methylation is actively involved in the down-regulation of cancer-associated genes in tumors, in particular, miRNA genes [19]. Therefore, we performed the analysis of methylation status of CpG islands of six cancer-associated miRNA genes (MIR-9- 1, MIR-9-3, MIR-132, MIR-148a, MIR-191, MIR-212) in 54 paired (tumor/normal) EOC samples.
The representative examples of MSP results for three miRNA genes (MIR-9-1, MIR-9-3, MIR-191) are shown in Figure 1. MSP products obtained with primers specific to methylated alleles were identified in a number of tumor samples and in several samples of histologically normal ovarian tissues from the same patients. In all samples of tumor and normal tissues, MSP products obtained with primers specific to unmethylated alleles were detected.
Table 2 summarizes the results of the methylation frequency analysis of six miRNA genes using the representative set of paired ovarian tissue samples from 54 patients with EOC. Methylation frequency was significantly higher in a group of tumor samples than in a group of matched normal samples for four miRNA genes: MIR-9-1, MIR-9-3, MIR-132, MIR- 148a (20-57 % vs 4-9 % (p < 0.01), respectively, by Fisher’s exact test). Besides, strong hypomethylation was revealed for MIR-191: 13 % vs 59 %, p ≤ 0.01. The most frequent hypermethylation in tumor samples was observed for MIR-9-1 (57 %), and hypomethylation of MIR-191 was also high (59 %). No changes were observed for MIR-212. These results suggest the involvement of aberrant methylation of five miRNA genes (MIR-9-1, MIR-9-3, MIR-132, MIR-148a, and MIR-191) in EOC initiation and development.
Hypermethylation of MIR-132 and MIR-148a, as well as hypomethylation of MIR-191 in EOC, were shown here for the first time.
Relationship between methylation frequency of examined miRNA genes and EOC progression
The data on methylation alterations of some miRNA genes obtained in the representative set of 54 EOC patients were correlated with clinical and histological characteristics of tumors. We revealed a significant dependence of methylation frequency of both MIR-9-1 and MIR-9-3 on a clinical stage, differentiation grade, and tumor size (p < 0.05, Fisher’s exact test, Figure 2). Besides, an association of MIR-9-1 and MIR-9-3 methylation frequency with the presence of metastases in lymph nodes and/or distant tissues was found on the level of a trend (p < 0.1, Fisher’s exact test, Figure 2). In addition, we revealed that the most significant methylation loss of MIR-191 was in low differentiated ovarian tumors (grade 3), namely, the frequency of MIR-191 methylation was equal to 3 % for tumor samples of grade 3 versus 22 % for tumor samples of grade 1 and 2 (p ≤ 0.05, Fisher’s exact test).
Up to our knowledge, this is the first report where a strong correlation of hypermethylation of MIR-9-1 and MIR-9-3 with a number of clinical characteristics, including advanced stage, grade and tumor size is shown, as well as with metastasis of ovarian cancer on the level of a trend. The association of hypomethylation of MIR- 191 with low differentiation of ovarian tumor cells was also shown here for the first time.
DISCUSSION
In this work, the search for novel miRNA genes with aberrant methylation in EOC was performed, and the association of hypermethylation or hypomethylation of examined miRNA genes with EOC progression was investigated. The methylation of six cancer-associated miRNA genes (MIR-9-1, MIR-9-3, MIR-132, MIR-148a, MIR-191, MIR-212) has been analyzed using a representative set of EOC samples.
The examined miRNAs play an important role in tumorigenesis of epithelial tumors. Five miRNA genes (excluding MIR-191) were suggested as typical tumor-suppressor genes and promising markers for prediction of cancer progression, in particular, ovarian cancer [10, 20-22]. MiR-132 was shown as a typical tumor-suppressor miRNA in EOC: expression level of miR-132 was dramatically decreased in ovarian cancer cell lines and clinical EOC tissue samples, considering that miR-132 can suppress cell proliferation, invasion, migration in ovarian cancer cells via targeting E2F5 [20]. On the contrary, it has been reported the enhanced expression level and proto-oncogenic behavior in ovarian cancer for miR-191 [23].
Previously, there were no publications concerning methylation of these genes in ovarian cancer, with the exception of a single previous report of hypermethylation of MIR-9-1 and MIR-9-3 as involved in drug resistance of EOC patients [10]. Nevertheless, data of this work on the tumor-suppressor role of miR-9 in EOC are in contradiction with another recent study that reported a possible proto-oncogenic function of miR-9 in ovarian tumors and its progression [24]. It was reported, that miR-9 overexpression was observed in EOC, and in this way, it may affect pathogenesis by targeting E-cadherin, thereby inducing an epithelial-mesenchymal transition [24]. These contradictions required further elucidation. Hypermethylation and hypomethylation are well-known markers that allow scientists to distinguish tumor-suppressor and proto-oncogenic features of any gene.
In the presented study, we revealed high frequency of hypermethylation of four out of six examined genes (MIR-9-1, MIR-9-3, MIR-132, MIR-148a) in EOC primary tumor samples in comparison with matched histologically normal samples: 20-57 % vs 4-9 %. These results are consistent with tumor-suppressor features of these genes, demonstrated earlier [20-22]. Besides, hypomethylation was revealed for MIR-191 (13 % vs 59 %), in agreement with the enhanced expression level of miR-191 and proto-oncogenic behavior in ovarian cancer [23].
Hypermethylation of MIR-132 and MIR-148a genes and hypomethylation of MIR-191 in EOC were reported here for the first time.
The absence of changes in the methylation status of MIR-212 in the tumors of these EOC patients is in agreement with a study suggesting histone modifications rather than DNA hypermethylation as epigenetic events, which regulate miR-212 levels in NSCLC [25].
Next, we correlated aberrant methylation frequencies of examined miRNA genes with clinical data on 54 ovarian tumors. It was shown a strong association of hypermethylation of MIR-9-1 and MIR-9-3 genes with clinical characteristics related to cancer progression: advanced III/IV clinical stages, low differentiation, large tumor size, and, on the level of a trend, presence of metastases in regional lymph nodes. Besides, we showed significant dependence of MIR-191 hypomethylation on the grade of ovarian cell differentiation.
In the case of down-regulation of miR-9 in ovarian cancer, it was reported to be associated with progression and drug resistance of EOC [10]. Our results show the relationship of MIR-9-1 and MIR-9-3 hypermethylation with advanced clinical stages, low grade of differentiation, large tumor size and metastasis, highlighting the role of miR-9 and hypermethylation of corresponding genes in progression of EOC. The data concerning the role of hypomethylation of MIR- 191 in EOC progression have been reported here for the first time.
Epigenetic markers for diagnosis and prognosis of the course of cancer attract more and more attention. We identified five miRNA genes (MIR-9-1, MIR-9-3, MIR-132, MIR-148a, MIR-191) with high frequency of aberrant methylation in EOC. Moreover, we tested four genes (MIR-9-1, MIR-9-3, MIR-132, MIR-148a) as a set of diagnostic markers. Indeed, these genes in total showed methylation in tumor samples with a frequency of 81.3 % (44/54) vs 20.4 % (11/54) in histologically normal samples. Thus, this combination of miRNA genes could have a screening discrimination value. In fact, it was shown that the frequencies of methylation alterations of MIR-9-1, MIR-9-3, and MIR-191 coincided with clinical characteristics of EOC, such as clinical stage, grade, tumor size, and metastasis presence.
In summary, our findings provide the evidence on the considerable contribution of aberrant methylation of five miRNA genes to ovarian cancer development (MIR-9-1, MIR-9-3, MIR-132, MIR-148a and MIR-191), and in MIR-9-1, MIR-9-3 and MIR-191 to ovarian cancer progression. Moreover, these results allowed us to suggest a novel potential biomarker system by using four of these miRNAs genes (MIR- 9-1, MIR-9-3, MIR-132 and MIR-148a) for early detection of EOC tumors with high sensitivity (81 % for the examined sampling) and specificity (80 % for the examined sampling).
ACKNOWLEDGEMENTS
This work was financially supported by the Russian Science Foundation (Project No. 14-15-00654).
REFERENCES
1. Engelberth SA, Hempel N, Bergkvist M. Development of nanoscale approaches for ovarian cancer therapeutics and diagnostics. Crit Rev Oncog. 2014;19(3-4):281-315.
2. Kinose Y, Sawada K, Nakamura K, Kimura T. The role of microRNAs in ovarian cancer. Biomed Res Int. 2014;2014:249393.
3. Coward JI, Middleton K, Murphy F. New perspectives on targeted therapy in ovarian cancer. Int J Womens Health. 2015;7:189-203.
4. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484-92.
5. Kunej T, Godnic I, Ferdin J, Horvat S, Dovc P, Calin GA. Epigenetic regulation of microRNAs in cancer: an integrated review of literature. Mutat Res. 2011;717(1-2):77-84.
6. Loginov VI, Rykov SV, Fridman MV, Braga EA. Methylation of miRNA genes and oncogenesis. Biochemistry (Mosc). 2015;80(2):145-62.
7. Piletic K, Kunej T. MicroRNA epigenetic signatures in human disease. Arch Toxicol. 2016;90(10):2405-19.
8. Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016;8(9):a019505.
9. Yang C, Cai J, Wang Q, Tang H, Cao J, Wu L, et al. Epigenetic silencing of miR-130b in ovarian cancer promotes the development of multidrug resistance by targeting colony-stimulating factor 1. Gynecol Oncol. 2012;124(2):325-34.
10. Li X, Pan Q, Wan X, Mao Y, Lu W, Xie X, et al. Methylation-associated Has-miR-9 deregulation in paclitaxel-resistant epithelial ovarian carcinoma. BMC Cancer. 2015;15:509.
11. Schmid G, Notaro S, Reimer D, Abdel-Azim S, Duggan-Peer M, Holly J, et al. Expression and promotor hypermethylation of miR-34a in the various histological subtypes of ovarian cancer. BMC Cancer. 2016;16:102.
12. Zuberi M, Khan I, Mir R, Gandhi G, Ray PC, Saxena A. Utility of serum miR-125b as a diagnostic and prognostic indicator and its alliance with a panel of tumor suppressor genes in epithelial ovarian cancer. PLoS One. 2016;11(4):e0153902.
13. WMA Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects [Internet]. Ferney-Voltaire: World Medical Association, Inc.; 2013 [cited 2016 Oct 11]. Available from: http://www.wma.net/en/30publications/10policies/b3/.
14. Sobin L.H. and Wittekind C. TNM Classification of Malignant Tumours, 6th edition. New York: Wiley; 2002.
15. Tavassoli FA, Devilee P. World Health Organization classification of tumours. Pathology and genetics of tumours of the breast and female genital organs. Lyon: IARC Press; 2003.
16. Pronina IV, Loginov VI, Burdennyy AM, Fridman MV, Kazubskaya TP, Dmitriev AA, et al. Expression and DNA methylation alterations of seven cancer-associated 3p genes and their predicted regulator miRNAs (miR-129-2, miR-9-1) in breast and ovarian cancers. Gene. 2016;576(1 Pt 3):483-91.
17. Loginov VI, Burdennyy AM, Pronina IV, Khokonova VV, Kurevljov SV, Kazubskaya TP, et al. Novel miRNA genes hypermethylated in breast cancer. Mol Biol (Mosk). 2016;50(5):797-802.
18. Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008;105(36):13556-61.
19. Vrba L, Munoz-Rodriguez JL, Stampfer MR, Futscher BW. miRNA gene promoters are frequent targets of aberrant DNA methylation in human breast cancer. PLoS One. 2013;8(1):e54398.
20. Tian H, Hou L, Xiong YM, Huang JX, Zhang WH, Pan YY, et al. miR-132 targeting E2F5 suppresses cell proliferation, invasion, migration in ovarian cancer cells. Am J Transl Res. 2016;8(3):1492-501.
21. Gong L, Wang C, Gao Y, Wang J. Decreased expression of microRNA-148a predicts poor prognosis in ovarian cancer and associates with tumor growth and metastasis. Biomed Pharmacother. 2016; 83:58-63.
22. Wei LQ, Liang HT, Qin DC, Jin HF, Zhao Y, She MC. MiR-212 exerts suppressive effect on SKOV3 ovarian cancer cells through targeting HBEGF. Tumour Biol. 2014;35(12):12427-34.
23. Dong M, Yang P, Hua F. MiR-191 modulates malignant transformation of endometriosis through regulating TIMP3. Med Sci Monit. 2015;21:915-20.
24. Yanaihara N, Noguchi Y, Saito M, Takenaka M, Takakura S, Yamada K, et al. MicroRNA Gene Expression Signature Driven by miR-9 Overexpression in Ovarian Clear Cell Carcinoma. PLoS One. 2016;11(9):e0162584.
25. Incoronato M, Urso L, Portela A, Laukkanen MO, Soini Y, Quintavalle C, et al. Epigenetic regulation of miR-212 expression in lung cancer. PLoS One. 2011;6(11):e27722.
Received in October, 2016.
Accepted in December, 2016.
Eleonora A Braga. Laboratory of Pathogenomics and Transcriptomics, Institute of General Pathology and Pathophysiology Moscow, 125315, Russia. E-mail: eleonora10_45@mail.ru.


{kind=link}
{kind=link}
{kind=link}
{kind=link}