










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN v.13 n.1 Santiago de Cuba ene.-feb. 2009
ARTÍCULO DE REVISIÓN
Hospital Infantil Sur
Enfermedades priónicas
Prion diseases
Tamara Rubio González 1 y Manuel Verdecia Jarque 2
RESUMEN
Las enfermedades priónicas son procesos neurodegenerativos producidos por el metabolismo aberrante de una proteína priónica, que afectan a seres humanos y animales durante un período de incubación prolongado, con carácter transmisible y evolución clínica fatal. Entre sus manifestaciones clínicas sobresalen: demencia, ataxia, insomnio, paraplejías, parestesias y conductas anormales. El principal hallazgo anatomopatológico es el aspecto espongiforme del cerebro de animales y personas infectados, causado por la acumulación de las proteínas priónicas en las neuronas, donde forman placas amiloides. No hay tratamiento que cure, mejore o controle los síntomas y signos de estas afecciones, por lo cual existen al respecto numerosas interrogantes y opiniones controvertidas en la comunidad científica mundial; razones todas que justificaron continuar polemizando en este artículo.
Descriptores:ENFERMEDADES POR PRIÓN,ENFERMEDADES POR PRIÓN/historia;ENFERMEDADES POR PRIÓN/transmisión, ENFERMEDADES POR PRIÓN/diagnóstico,ENFERMEDADES POR PRIÓN/etiología;SÍNDROME DE CREUTZFELDT-JAKOB;KU
RU;ENFERMEDAD DE GERSTMANN-STRAUSSLER-SCHEINKER;INSOMNIO FAMILIAR FATAL
Límites: HUMANO; ANIMAL
ABSTRACT
Prion diseases are neurodegenerative processes occurred by aberrant metabolism of a prion protein that affect humans and animals during a long period of incubation, with transmissible character and fatal clinical course. Among their clinical manifestations are insanity, ataxia, insomnia, and paraplegias, paresthesias and abnormal behaviors. The main patological finding is the spongiform aspect of the infected animal and human brain caused by accumulation of prion proteins in the neurons, where they form amyloid plaques. There is not treatment that cures, improves or controls symptoms and signs of these conditions, therefore several questions and different opinions in this regard raise in the world scientific community that justified to continue arguing in this paper.
Subject heading:PRION DISEASES,PRION DISEASES/history;PRION DISEASES/transmission,PRION DISEASES/diagnosis,
PRION DISEASES/etiology;CREUTZFELDT-JAKOB SYNDROME;KURU;GERSTMANN STRAUSSLER-SCHEINKER DISEASE;
INSOMNIA, FATAL FAMILIAL
Limits: HUMAN; ANIMAL
Recibido: 13 de mayo del 2008
Aprobado: 10 de junio del 2008
Las enfermedades priónicas constituyen el nuevo paradigma en la nosología neurológica. Aunque hasta ahora su incidencia es baja, su carácter transmisible plantea nuevos problemas de salud pública. 1,2
Pueden definirse como entidades neurodegenerativas que afectan a humanos y animales, producidas por el metabolismo aberrante de una proteína priónica (PrP), que presentan un período de incubación prolongado, transmisibilidad y evolución clínica fetal. Se ha utilizado el término de prionpatías para nombrarlas y debido a la espongiosis que producen en el sistema nervioso también se les conoce con el nombre de encefalopatías espongiformes subagudas. 1, 3
La historia de estas enfermedades recoge, como primeras referencias, la descripción que hicieran en el siglo XVIII los ganaderos europeos de una enfermedad neurodegenerativa total que afectaba a ovejas y cabras, denominadas tembladera (scrapie, en inglés) 4 Llama la atención el aspecto de esponja del cerebro de estos animales, de donde se deriva el término de espongiforme. No fue hasta principios del siglo XX que se describieron por Creutzfeldt y Jakob los primeros casos de encefalopatía espongiforme en el hombre, por lo que la enfermedad recibió la denominación de enfermedad de Creutzfeldt-Jacob. 5 En 1960 el grupo de Gajdusek 6 demostró su transmisibilidad y en 1982 Stanley Prusiner descubrió el agente patógeno, el prión, demostrando que se trataba de partículas puramente proteicas sin ácido nucleico, trabajo por el cual le fue otorgado el Premio Nobel de Medicina en 1997. 3
Estos trabajos descubren la existencia de dos proteínas PrP distintas, una de ellas causa la enfermedad. Ambas poseen igual secuencia, pero distinta conformación tridimensional. Además, las PrP anormales o patógenas parecen ser capaces de inducir el replegamiento de las PrP normales. 3, 7 Estos hallazgos causaron gran desconcierto en la comunidad científica debido a su clara contradicción con el dogma central de la biología: todas las formas de vida, desde los virus hasta las plantas y los animales superiores, transmiten sus caracteres a las siguientes generaciones a través del ADN (excepcionalmente ARN). El flujo de la información es en todos los casos: ADN " ARNm " secuencia de aminoácidos " estructura tridimensional de la proteína.
La teoría de los priones supone la existencia de dos plegamientos para una única secuencia de aminoácidos y lo que resulta más interesante e inquietante, el replegamiento de la PrP normal por acción de la PrP patológica, sugiere un flujo de información de una proteína a otra a nivel de estructura terciaria. 3, 4, 7 Por tal motivo los priones constituyen las únicas partículas vivas que contradicen el dogma central de la biología.
Propiedades del prión o proteína priónica
El prión o proteína priónica es una partícula acelular, patógena y transmisible y posee la propiedad de desnaturalizar otras proteínas. Teorías más recientes sugieren que los priones son proteínas modificadas bajo circunstancias que favorecen su caída a un nivel energético muy estable, confiriéndole nuevas propiedades biológicas, tales como ser insolubles, resultar inmunes a las proteasas y cambiar su configuración tridimensional. 7, 8
Además cuentan entre sus propiedades las siguientes:
§ No contiene ADN ni ARN.
§ Carece de cuerpos de inclusión.
§ Período de incubación prolongado (meses, años, décadas).
§ No ocasiona respuesta inflamatoria.
§ No genera respuesta antigénica.
§ Curso crónico progresivo.
§ Invisible al microscopio electrónico.
La falta de respuesta inmunitaria a las infecciones por priones no implica que estas proteínas eludan el sistema inmunológico, sino que por el contrario, lo utilizan en las etapas iniciales de acumulación y replicación priónica. Los últimos indicios al respecto apuntan a que los priones se acumulan y se replican, inicialmente en las células dendríticas foliculares (FDC) de los centros germinales y que las células B están implicadas en la neuroinvasión, fundamentalmente, mediante su participación en la maduración de las FDC. 9
El gen PRNP
La proteína priónica está codificada por un gen denominado PRNP y se localiza en 20 p12 – pter, el locus es pter – PRP – SCG1 (figura 1). Contiene 15 000 pares de bases y se encuentra altamente conservado. Ha sido identificado en más de trece especies de mamíferos. Está compuesto por dos exones y un intrón. El extremo 5´ del sitio de inicio de la transcripción presenta alta repetición de pares GC. 7, 10

Figura 1. Brazo corto del cromosoma 20
En ovejas se conocen ocho alelos para PRNP. Cada uno resulta de una mutación puntual y su frecuencia varía según raza y rebaño. El alelo PrPMARH se asocia con elevadas incidencias de scrapie. La secuenciación del gen en ovejas ha puesto de manifiesto la existencia de regiones homólogas en los genes ovino y humano.
En hamsters el gen PRNP contiene dos exones separados por un intrón de 10 Kb. El exón 1 codifica una porción de la secuencia líder 5´, y el exón 2 codifica la proteína PrP y la secuencia no traducida 3´.
En ratones el gen PRNP está compuesto por tres exones, el exón 3 es análogo al exón 2 del hamster. La localización de los genes PRNP en el brazo corto del cromosoma 20 humano y en una región homóloga del cromosoma 2 del ratón sugirió que estos genes existían antes de la especiación de los mamíferos. 11
En el hombre se han registrado más de 20 mutaciones, resultado de deleciones o inserciones de una o más bases nucleotídicas, en la región codificadora del gen PRNP en todas las formas familiares de prionpatías; sin embargo, en las formas esporádicas de estas enfermedades no se han demostrado mutaciones en el gen. 2, 7
Proteínas criónicas
El producto proteico del gen PRNP es una proteína celular (PrPc) proteasa sensible que se acumula especialmente en la membrana citoplasmática de las neuronas y tiene entre 33 – 35 KD. Está constituida por una sola cadena peptídica, presenta una estructura compacta de cuatro hélices alfa (H1 – H4) y oligosacáridos complejos unidos a la proteína. La composición total incluye 2% de hélices alfa y 3% de hélices beta. 8, 11
La PrPc consta de 253 amoniácidos en el hombre y 254 en ratones y hamster, está muy conservada en diversas especies que incluyen humanos, ovejas, bóvidos, ratones, hamsters y Drosophila. Se encuentra anclada a la membrana neuronal por un glicosyl – fosfatidilinositol (GPI). Además está ampliamente expresada en el organismo (bazo, músculo esquelético, pulmones y epitelios secretores) aunque con niveles muy inferiores al cerebro. Se sintetiza en el retículo endoplásmico rugoso, es modificada en el aparato de Golgi y luego transportada a la superficie celular mediante vesículas secretoras.
La forma patógena de PrP (PrPsc), a diferencia de PrPc, presenta gran proporción de hélices beta (43% de hélices beta y 30% de hélices alfa). 8,11
La estructura tridimensional de la proteína ha sido estudiada por resonancia magnética nuclear (RMN), pues no ha sido posible producir cristales de esta en el laboratorio. De tal forma pudo determinarse la presencia de un extremo N – terminal flexible (a.a 23 – 124), un dominio globular central (a.a 125 – 228) y una cadena C – terminal corta (a.a 228 – 230 o 231). El dominio globular contiene tres hélices alfa y a cada lado de la segunda hélice se encuentra una lámina beta corta. La transformación de PrPc en PrPsc implica probablemente el replegamiento de la región que incluye los aminoácidos 90 – 140, que se transforman en láminas beta (figura 2). Esta enorme proporción de hélices beta conduce a la formación de fibrillas o bastoncillos amiloides denominados SAF (Scrapie Associated Fibrils) que se encuentran en el cerebro de las personas y animales enfermos. 12
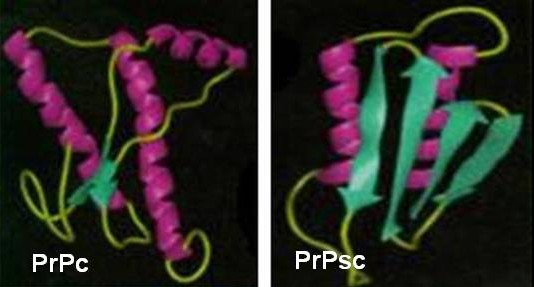
Figura 2. Formas de la proteína priónica
Conversión de PrPc en PrPsc
Aunque ambas formas proteicas, PrPc y PrPsc son codificadas por el gen PRNP, no pueden ser el resultado de un procesamiento alternativo del ARNm. Para explicar la conversión de PrPc en PrPsc se han propuesto diversas hipótesis. 10-13
§ Según la teoría de Prusiner, hay una interacción directa entre una molécula PrPc y una PrPsc. Esta última induce la conversión de la primera en una segunda molécula PrPsc (figura 3), sin embargo, aún no se han detectado agregados PrPc - PrPsc.
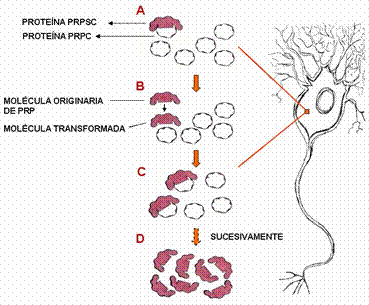
Figura 3. Conversión de PrPc en PrPsc según la teoría de Prusiner
§ El modelo de semilla o núcleo propone que la transformación podría ser el resultado de una polimerización en cadena, iniciada a partir de la inoculación de PrPsc. La semilla consta de 6 unidades de PrP (figura 4).
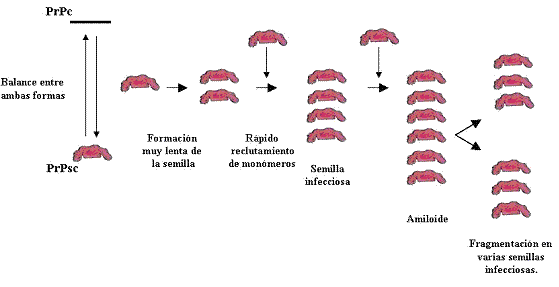
Figura 4. Modelo de semilla
§ Transformación química postraduccional de PrPsc. Tal hipótesis concuerda con la acumulación lenta de PrPsc en los cerebros de animales y hombres infectados, a pesar de los niveles invariables de ARNm durante la evolución de la enfermedad.
§ Existencia de elementos ajenos a PrPc que, en presencia de PrPsc, interfieren con la proteína anormal e inducen su cambio conformacional.
§ La teoría del holoprión de Weissmann propone que la conversión puede ser mediada por un holoprión o por la unión de un coprión a PrPc. Esta modificación implica alteraciones químicas y conformacionales.
§ La intervención de chaperonas (proteína X) que modifican el plegamiento de PrPc o de su precursor (considerando que PrPsc puede ser incluso la chaperona de PrPc).
§ Conversión preventiva por barrera de energía (figura 5). Esta teoría propone que bajo ciertas circunstancias PrPc modifica su nivel energético para lograr estabilidad, lo que provoca un cambio en su configuración tridimensional que la hace insoluble y muy resistente a la degradación por proteasas.

Figura 5. Conversión preventiva por barrera de energía
A pesar de las numerosas hipótesis queda claro que un punto fundamental a resolver es impedir que la proteína PrPc normal se transforme en PrPsc patológica, meta que parece aún lejana, pero el debate vuelve a intensificarse al Chiesa y Harris 14 plantear lo siguiente:
1. Se ha comprobado la presencia de PrPsc sin síntomas clínicos ni alteraciones histopatológicas de encefalopatía espongiforme aguda.
2. Ocasionalmente se ha observado el cuadro clínico y el complejo neuropatológico de encefalopatía espongiforme subaguda con PrPsc escasa o indetectable de ratones inoculados con scrapie y con encefalopatía espongiforme bovina.
3. Ratones inoculados con priones de hamster adquieren niveles altos de PrPsc, degeneración esponjosa y placas amiloides en cerebro, pero no expresan síntomas clínicos.
4. En enfermedades priónicas familiares parece no ser necesaria la presencia de PrPsc para desarrollar la enfermedad.
Después de estas evidencias los biólogos moleculares se preguntan: ¿podría la PrPc inducir neurodegeneración sin convertirse en PrPsc?, y ¿podría la alteración en el plegamiento de proteínas ser parte de un proceso patológico, pero también fisiológico o normal?
Puede ser lógico pensar que bajo ciertas circunstancias celulares, que impliquen situaciones de estrés celular, capaces de llevar a un desbalance energético, la PrPc modifica su nivel energético para ser más eficiente y mantener su estabilidad, ya que la capacidad de guardar información conformacional de los priones los convierten en candidatos para participar en procesos celulares que demandan estabilidad durante largos períodos (formación de la memoria a largo plazo, la memoria inmunológica y la evolución del genoma de muchos organismos). Debido a que los priones son menos susceptibles a la digestión de las proteasas es muy probable que se trate de mecanismos celulares fisiológicos.
La barrera de especie
La barrera de especie para los agentes infecciosos radica en la secuencia de aminoácidos de la PrPc normal. Esta evidencia justificaría el hecho de que vacas inglesas desarrollaron encefalopatía espongiforme bovina al consumir tejidos de ovejas afectadas de scrapie: las proteínas PrP de estas especies difieren tan solo en 7 posiciones. Sin embargo, las proteínas PrP de vacas y humanos difieren en más de 30.
En la actualidad se acepta que la enfermedad de Creutzfeldt – Jakob es el homólogo humano de la encefalopatía espongiforme bovina (EEB). Existe la hipótesis de que esta última es transmitida al hombre por la ingestión de animales infectados (vacas y ovejas). Se ha demostrado que el agente de la EEB puede ser transmitido de primate a primate. La misma cepa de EEB causante de la enfermedad en bovinos está implicada en la ocurrencia de encefalopatía espongiforme en gatos domésticos, tigres y algunos rumiantes en zoológicos. Se transmite por inyección parenteral de cerebro infectado a ovejas, cabras, cerdos, visón, micos titíes y ratones, pero no a hamsters o pollos. 15- 18
Todas estas evidencias sugieren que los priones han superado la barrera de especie.
Dianas para la infección por priones
Según el grado de infecciosidad de la Organización Mundial de la Salud serían: 19
- De alto riesgo: cerebro, hipófisis, médula espinal, bazo, duramadre, timo, amígdalas, placenta, ojos, ganglios linfáticos e intestinos.
- De riesgo moderado o bajo: nervios periféricos, líquido cefalorraquídeo, páncreas, hígado, glándula suprarrenal, pulmón, médula ósea y músculo esquelético.
- No relacionado con infectividad en ninguna especie: leche, saliva, piel, semen, orina, sangre, heces, riñón y huesos.
Clasificación de las enfermedades priónicas humanas
La presente clasificación se deriva de la historia de estas enfermedades y refleja las incógnitas que aún persisten sobre estas, por lo que tiene carácter provisional. 20- 22
§ Esporádicas:
- Enfermedad de Creutzfeldt - Jacob
· Clásica
· Variante de Heidenhain
· Variante de Brownell – Oppenheimer
· Panencefalopatía
§ Adquiridas:
- Kuru (canibalismo)
- Enfermedad de Creutzfeldt – Jacob iatrogénica
· Hormona de crecimiento
· Gonadotropina
· Trasplante de córnea
· Electrodos de EEG de implantación directa
· Implante de duramadre
· Neurocirugía?
- Nueva variante británica (¿EEB?)
§ Familiares:
- Enfermedad de Creutzfeldt – Jacob familiar
- Síndrome de Gerstmann – Sträussler – Scheinker
- Insomnio familiar fatal
- Enfermedades por priones atípicas
Manifestaciones clínicas de las enfermedades priónicas humanas
El espectro de manifestaciones clínicas de estas enfermedades es amplio. Cada una de ellas parece estar relacionada con una conformación específica de
El período de incubación puede alcanzar 30 o más años. 23 Los pacientes pueden presentar demencia, ataxia, insomnio, paraplejias, parestesias y conductas anormales. 24 - 31 Puede manifestarse un declive progresivo de las funciones cognitivas y motoras.
Enfermedad de Creutzfeldt – Jacob (ECJ)
Es la enfermedad priónica más común. Se tienen referencia de casos en todo el mundo y su prevalencia se calcula cercana a 1:1000 000 000 de habitantes. El debut aparece entre los 50 y los 60 años y su curso clínico puede tener varias fases hasta alcanzar una demencia grave con mioclonias. En la mayoría de los pacientes el deterioro mental es lentamente progresivo con trastornos cognoscitivos, pérdida de la memoria y delirio. La triada clínica incluye demencia progresiva, mioclonias y EEG con complejos períodos trifásicos de 1-2 Hz. Pueden apreciarse cambios en el comportamiento, disfunción cortical alta y alteraciones visuales (variante de Heidenhain), signos de alteración cerebelosa (variante de Brownell-Oppenheimer), manifestaciones piramidales, extrapiramidales y disfunción de neurona motora inferior. En fases avanzadas de la enfermedad puede aparecer mutismo aquinético. La muerte sobreviene entre 1 mes a 10 años, con una media de 1 año después del debut clínico. 32- 34
La forma familiar de la enfermedad (ECJf) representa de 10 a 15% de personas con ECJ y sigue una herencia autosómica dominante. La mutación más frecuentemente registrada en todo el mundo es E200K que implica una sustitución de glutamato (E) por lisina (K) en el codón 200. ECJf debuta, por lo general, más tempranamente, tiene un curso más rápido y mayor postración. La mutación E200K se caracteriza por determinar la aparición temprana de pérdida de memoria y confusión que luego se acompañan de demencia rápidamente progresiva, signos piramidales y extrapiramidales, ataxia mioclonias. También se ha informado con esta mutación la ocurrencia de oftalmoplejia supranuclear y neuropatía periférica desmielinizante.
Los hallazgos anatomopatológicos consisten en atrofia cerebral y cerebelosa, vacuolización del neuropilo en la substancia gris con disminución de la población neuronal, astrocitosis marcada, placas amiliodes y ausencia de infiltrado inflamatorio. 35
Enfermedad de Gertsmann – Straussler – Scheinker
Se caracteriza por una ataxia grave acompañada de paraparesia espástica. Su inicio puede ser tan temprano como a los 20 años, pero puede aparecer hasta los 60 años. La duración es entre 2 y 10 años y los pacientes fallecen generalmente por infecciones secundarias. 23, 31 La mutación más frecuentemente registrada es en el codón 102, que determina un cambio de prolina a leucina. También se han encontrado mutaciones en los codones 105, 145 y 117. Sigue una herencia autosómica dominante.
El estudio anatomopatológico evidencia varios tipos de placas focales o difusas en la corteza cerebral y cerebelosa, que resultan ser inmunorreactivas a los anticuerpos anti PrP humanos. Además aparece degeneración espinocerebelosa.
Kuru
El término Kuru significa en lengua aborigen temblor, con fiebre y frío. Fue inicialmente descrita en la tribu Papúa de Nueva Guinea y aparecía después de la ingestión de tejidos cerebrales de personas fallecidas con la finalidad de adquirir su sabiduría. Predomina en niños y mujeres adultas.
La enfermedad se desarrolla lentamente, y el período de incubación puede prolongarse hasta 30 años. Progresa usualmente hasta la muerte en aproximadamente 1 año. Gajdusek descubrió tres etapas en su curso clínico:
§ Fase ambulatoria: aparece temblor generalizado con pérdida de la capacidad para coordinar los movimientos y disartria, lo cual evidencia una afectación cerebelosa incipiente.
§ Fase sedentaria: se manifiesta incapacidad de deambulación independiente, temblores más fuertes, ataxia, labilidad emocional, depresión y bradipsiquia. Están conservados los reflejos tendinosos y aún no es perceptible la degeneración muscular.
§ Fase terminal: hay incapacidad para la sedestación independiente, ataxia severa, temblores, disartira, incontinencia urinaria y fecal, disfagia y ulceraciones cutáneas. 36- 39
Está caracterizada por extensa vacuolización, gliosis, infiltrado inflamatorio mínimo o ausente y placas amiloides.
Insomnio familiar fatal (IFF)
Constituye una variedad rara de enfermedad priónica. Su aparición tiene lugar entre los 35 - 55 años, con una edad media de inicio a los 45 años. El tiempo de vida media una vez que comienza clínicamente la enfermedad es de 1 año. Ha sido descrita la mutación en el codón 178 que provoca cambios en metionina y valina. Se caracteriza por un insomnio intratable que dura semanas o meses. Aparece seguidamente disautonomía, ataxia y signos piramidales y extrapiramidales. Las funciones cognoscitivas se conservan relativamente hasta períodos avanzados de la enfermedad. Estos pacientes pueden presentar alteración episódica de la tensión arterial, la frecuencia cardíaca y respiratoria y de la temperatura. En el EEG puede encontrarse un enlentecimiento difuso con más frecuencia que descargas periódicas. 40, 41
La neuropatología de esta enfermedad incluye anomalías en tálamo y oliva bulbar inferior. En el tálamo se puede apreciar pérdida marcada o subtotal de neuronas, especialmente en los núcleos anterior y dorso medial, asociada con astrogliosis. No se observa espogiosis en el tálamo, pero pueden encontrarse focos aislados en la corteza cerebral y la sustancia blanca subyacente, fundamentalmente en áreas límbicas y en la capa de células de Purkinje del cerebelo.
Hay notable pérdida de células de Purkinje en cerebelo. En resumen, a nivel neuropatológico, se puede considerar una degeneración tálamo – olivar con cambios corticales leves, especialmente límbicos, en relación con la duración de la enfermedad. 26
En la tabla se muestran las principales manifestaciones clínicas de algunas enfermedades priónicas humanas, así como la edad de inicio de los síntomas y la duración.
Tabla. Manifestaciones clínicas de las enfermedades criónicas
| Tipo | Síntomas | Edad | Duración |
| Kuru | § Ataxia | 40 años | 3 meses – 1 año |
| Enfermedad de Creutzfeldt – Jacob | § Demencia | < 60 años | 1 mes a 10 años (media 1 año) |
| Enfermedad de Gerstmann – Sträuler - Scheinker | § Ataxia | < 60 (20-60) | 2 a 10 años |
| Insomnio familiar letal | § Insomnio | 45 ± 10 | 1 año |
Tratamiento
La comunidad científica dirige sus esfuerzos para encontrar un medicamento que cumpla con el principio farmacológico de traspasar la barrera hematoencefálica y sea capaz de impedir la conversión de la proteína priónica normal en proteína anormal. Con estos fines han sido utilizados ciertos tipos de medicamentos ya conocidos en el tratamiento de enfermedades neurológicas, estos son derivados de la quinacrina y la clorpromazina, utilizados en el tratamiento de la malaria y la esquizofrenia, respectivamente.
Estudios recientes in vitro han indicado que los anticuerpos monoclonales anti-PrP, con escasa o nula afinidad por la PrPsc, pueden prevenir la incorporación de PrPc a los priones infectantes. Igualmente estos estudios mostraron un efecto similar in vivo. 42, 43 Estos hallazgos son esperanzadores y señalan a las estrategias inmunoterapéuticas en humanos como una posibilidad de tratamiento para las enfermedades por priones.
En consecuencia, no existe actualmente un tratamiento que cure, mejore o controle las manifestaciones clínicas de estas enfermedades. Además los ensayos con las drogas mencionadas y con anticuerpos monoclonales en modelos animales, se han probado la amantadita, los esteroides, el interferón, el aciclovir, diversos agentes antivirales y antibióticos; sin embargo, ninguno ha demostrado claramente su eficacia. 12
Similitud con otras enfermedades neurodegenerativas:
En algunos estudios se ha encontrado similitud de estas afecciones con la enfermedad de Parkinson, la esclerosis lateral amiotrófica y la enfermedad de Alzheimer. Esta relación se basa en que en estas enfermedades hay casos esporádicos y familiares. 4, 19
En la enfermedad de Creutzfeldt – Jacob y en la de Alzheimer aparece un cambio en la configuración espacial de proteína priónica en la primera, y en la beta – amiloide en la segunda. Se ha observado también una variante vascular de la proteína priónica, la presencia de ovillos neurofibrilares intraneuronales como fenotipo de una mutación en el codón 145 del gen PRNP. En la enfermedad de Alzheimer, la alipoproteína E podría incidir como un chaperón molecular respecto a la proteína A4 de la beta – amiloide.
CONCLUSIONES
Todavía constituye una controversia el hecho de que una proteína provoque una enfermedad infecciosa. Otro punto sin respuesta es el mecanismo por el cual el agente infeccioso se multiplica en el individuo afectado si no contiene ácidos nucleicos, y esta misma característica genera otra interrogante no resuelta, relacionada con la memoria genética: ¿cómo puede la proteína priónica guardar la información sin alterar los aminoácidos que la forman?, simplemente moldea o cambia su forma por mecanismos desconocidos aún.
También es interesante tener en cuenta que una proteína que se expresa en el cerebro y otros tejidos durante el desarrollo embrionario, en todos los mamíferos, puede ser prescindible sin efectos negativos.
Las respuestas a estas interrogantes constituyen hoy un desafío a la comunidad científica.
REFRENCIAS BIBLIOGRÁFICAS
1. Zarranz JJ.Enfermedades priónicas o prionpatías.Neurología 2006;21(8):396–399.
2.Hernández FAA,Céspedes CG,González AJE.Enfermedades priónicas en humanos.Gac Med Caracas 2002;110(1).
<http://www. anm.org>[consulta:16 febrero 2008].
3.Prusiner SB.Prion diseases and the BSE crisis.Science 1997;278:245– 51.
4.Pastor ST.Priones,solución a un enigma médico.Seminarios de Biología Celular 1999.<http://www.retina.umh.es/docencia/bioce
lular/seminarios/seminario/priones1.html>[consulta:9 mayo 2008].
5.Polo JM.Historia y clasificación de las enfermedades priónicas humanas.Foro de trostemas neurológicos 1998.<http://www.svneu rologia.org/congreso/priones-3.html>[consulta:20 septiembre 2007].
6.Gajdusek DC,Gibas CJ,Alpers M.Experimental transmision of a Kuru-like syndrome to chimpanzes.Nature 1996;209:794–96.
7.Gasset M,Westaway D.Los priones y su biología 1998.<http://www.neurologia.rediris.es/congreso1/conferencias/priones.html>
[consulta:3 mayo 2008].
8.Wikimedia Foundation,Inc.Prion 2007.<http://www.es.wikipedia.org/wiki/Pri%C3%B3n>[consulta:2 febrero 2008].
9.Ogayar SA,Sánchez PM.Variabilidad conformacional en las encefalopatías espongiformes transmisibles.El sistema inmunitario
en las enfermedades priónicas.2007.<http://www_grupoaran_com_ediciones.htm>[consulta:2 marzo 2008].
10.Oesch B,Westaway D,Walchli M,McKinley MP,Kent SB,et al.A cellular gene encodes scrapie Prp 27–30 protein.Cell 1985;40:
735–46.
11.Oesch B.Un gen, dos proteínas.En:Prion Review.Chapter IV.2007.<http://www.biologia.educar/el_prion/prion4.htm>[consulta:16 marzo 2008].
12.López-Herrera A,Haenni AL,Ureuqui–Inchima S.El desafío de las enfermedades priónicas,una emergencia en humanos y bovi
nos.Vet Méx 2002; 33(4).
13. Weissmann C.The prion`s progress.Nature 1991;349:569–71.
14.Chiesa R,Harris DA.Prion Diseases:What is the neurotoxic molecule?Neurobiology of Disease 2001; 8: 743–763.
15.Patterson WJ,Painter MJ.Bovine spongiform encephalopathy and new variant Creutzfeldt–Jakob disease:an overview.Commun Dis Public Health 1999;2:5–13.
16.Fishbein L.Transmissible spongiform encephalopathies,hypotheses and food safety:an overview.Sci Total Environ 1998;217:9871–82.
17.Narang lt.Origin and implications of bovine spongiform encephalopathy.Proc Soc Exp Biol Med 1996; 211:306–22.
18.Collee JG,Bradley R.BSE:a decade on–Part 1.Lancet 1997; 49:636–41.
19.Mora FJ.Cuestiones sobre las enfermedades priónicas.Rev Neurol 2000;31(2):129-70<http://www.revneurol.com/3102.htm>[consulta:16 febrero 2008].
20.Silva R.Human spongiform encephalopathy.Clinical presentation and diagnostic test.En:Baker HF y Ridley RM,eds.Prion diseases.Totowa:Humana Press 1996: 15 – 33.
21.Ridley RM,Baker HF.Fatal protein.The story of CJD,BSE,and other prion diseases.Oxford:Oxford University Press 1998.
22.Gimeno AA.Las enfermedades del futuro:el reto de los priones.2001.<http://www.medynet.com/elmedico/documentos/anuario 01/indice.pdf>[consulta:16 febrero 2008].
23.Prusiner SB.El prión en la patología.Investigación y ciencia.<http://coli.usal.es/web/educativo/articulos/art11/Fig02.gif>
[consulta:3 abril 2008].
24.Ayuso T,Muñón T,Erro ME.Patología del sueño en las enfermedades priónicas.An Sist Sanit Navar 2007;30(Supl):135–41.
25.McKintosh E,Tabrizi SJ,Collinge J.Prion diseases.Journal of Neurovirology 2003;9:183-93.
26.Montagna P.El insomnio familiar fatal:aspectos clínicos,de laboratorio y patológicos.Rev Neurol 1999;29:1006–9.
27.Ayuso T,Urriza J,Caballero C,Iriarte J,Muñoz R,García–Bragado F.Insomnio letal familiar:estudio clínico,neurofisiológico e histopatológico de dos casos.Neurología 2006;21:414-20.
28.Meissner B,Köhler K,Körtner K,Barth M,Jastrow U,Mollenhaver B,et al.Sporadic Creutzfeldt–Jakob disease.Magnetic resonance imaging and clinical findings.Neurology 2004; 63: 450-56.
29.Landott H,Glatzel M,Blattler T,Achermann P,Roth C,Mathis J.Sleep–wake disturbances in sporadic Creutzfeldt–Jakob disease.Neurology 2006;66:1418-24.
30.Mora FJ.Cuestiones sobre las enfermedades priónicas<http://www.neurovia.org/700/eprionicas.htm>[consulta:3 abril 2008].
31.Alom J.Clínica de las enfermedades priónicas humanas 1998.<http://suneurologia.org/congreso/priones-5-html>[consulta:3 abril 2008].
32.Kretzchmar HA,Ironside JW,De Armond SJ,Tateishi J.Diagnostic criteria for sporadic Creutzfeldt–Jakob disease.Arch Neurol 1996;53:913–20.
33.Will RG,Zeidler M,Stewart GE,Macleod MA,Ironside JW,Cousens SN,et al.Diagnosis of new variant Creutzfeldt–Jakob disease.Ann Neurol 2000;47:575–82.
34.Hernández A,Céspedes G,González A,Álvarez A,Lara C,Soto A,et al.Enfermedad de Creutzfeldt–Jakob en Venezuela:informe de 10 casos y revisión de la literatura.Rev Inst Nac Hig Rafael Rangel 1999; 30:14 –20.
35.Ironside JW,Head MW,Bell JE,Mc Cardle L,Will RG.Laboratory diagnosis of variant Creutzfeldt–Jakob disease.Histopatology 2000;37:1-9.
36.Gajdusek DC.Krum:From the New Guinea field journals 1957–1962.Grand Streel 1996;15:6-33.
37.Prusiner SB.Prion diseases.Scientific American 1995;15:6–33.
38.Collinge J,Whitfield J,McKintosh E,Beck J,Mead S,Thomas D,AlpersM.Kuru in the 21st century:an acquired human prion disease with very long incubations periods.Lancet 2006;367:2068-74.
39. Zigas V.Laughing death.The untold store of Kuru.Clifton:Human Press,1970.
40.Montagna P,Gambetti P,Cortelli P,Lugaresi E.Familial and sporadic fatal insomnia.Lancet Neurol 2003; 2:167-76.
41.Montagna P.Fatal familiar insomnia:a model disease in sleep physiopathology.Sleep Med Rev 2005;9: 339 –53.
42.Korth C,May BCH,Cohen FE,Prusiner SB.Aeridine and phenothiazine derivates as pharmacotherapeutics for prion disease.
Proc Natl Acad Sci USA 2001;98:9836–41.
43.White AR,et al.Monoclonal antibodies inhibit prion replication and delay the development of prion disease.Nature 2003;422:
80–3.
Dra. Tamara Rubio González. Calle 7, no. 33A altos, entre 1ra y 2da. Reparto Ampliación de Fomento, Santiago de Cuba.
1 Especialista de II Grado en Genética Clínica
Hospital Infantil Sur Docente, Santiago de Cuba, Cuba
2 Especialista de I Grado en Oncología
Hospital Infantil Sur Docente, Santiago de Cuba, Cuba
CÓMO CITAR ESTE ARTÍCULO
Rubio González T,Verdecia Jarque M.Enfermedades criónicas.[artículo en línea]MEDISAN 2009;13(1).<http://bvs.sld.cu/revistas/san/ vol13_1_09/san08109.htm>[consulta: fecha de acceso].

{kind=link}