










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN vol.15 no.9 Santiago de Cuba set. 2011
ARTÍCULO ORIGINAL
Síndrome de Usher de tipo II: caracterización oftalmológica, auditiva y genética de una familia consanguínea
Type II Usher syndrome: ophthalmological, auditory, and genetic characterization of a consanguineous family
MsC. Rásife Freyre Luque, 1 MsC. Sarah María García Espinosa, 2 MsC. Idalmis García Mayet, 3 Dra. Francisca Santisteban Aguilera 4 y MsC. Melek Dager Salomón 5
1 Especialista de I Grado en Oftalmología. Máster en Medicina Bioenergética y Natural. Profesora Asistente. Centro Provincial de Retinosis Pigmentaria, Santiago de Cuba, Cuba.
2 Especialista de II Grado en Oftalmología. Máster en Medicina Bioenergética y Natural. Profesora Asistente. Investigadora Agregada. Centro Provincial de Retinosis Pigmentaria, Santiago de Cuba, Cuba.
3 Especialista de I Grado en Oftalmología y en Medicina General Integral. Máster en Medicina Bioenergética y Natural. Instructora. Centro Provincial de Retinosis Pigmentaria, Santiago de Cuba, Cuba.
4 Especialista de II Grado en Otorrinolaringología. Profesora Auxiliar. Hospital Provincial Docente Clinicoquirúrgico "Saturnino Lora Torres", Santiago de Cuba, Cuba.
5 Especialista de I Grado en Genética. Máster en Atención Integral al Niño. Profesora Asistente. Hospital Infantil Sur, Santiago de Cuba, Cuba.
RESUMEN
Se caracterizó a una familia consanguínea de 25 miembros, 3 de los cuales padecían el síndrome de Usher de tipo II, a través del estudio auditivo, oftalmológico y genético en el Centro de Retinosis Pigmentaria de Santiago de Cuba. Los pacientes (2 varones y 1 fémina) tenían en común: aparición de la enfermedad en la etapa juvenil, mala visión nocturna, campos visuales reducidos, hipoacusia neurosensorial y resultados normales en las pruebas vestibulares; asimismo, en genética molecular, la electroforesis en gel de poliacrilamida reveló la presencia del marcador D1S237, estrechamente ligado al gen USH2 en el cromosoma 1. Esa caracterización permitirá aplicar la terapia génica y los implantes, tanto de células madre como cocleares, según corresponda.
Palabras clave: familia, síndrome de Usher, hipoacusia, campo visual, genética, retinosis pigmentaria.
ABSTRACT
A consanguineous family of 25 members, 3 of whom suffered from type II Usher syndrome was characterized through the auditory, ophthalmologic, and genetic study in the Retinitis Pigmentosa Center from Santiago de Cuba. The patients (2 males and a female) had in common: occurrence of the illness during youth, bad night vision, reduced visual fields, neurosensorial hypoakusia, and normal results in the vestibular tests; also, in molecular genetics, electrophoresis in polyacrilamide gel revealed the presence of the D1S237 marker, closely linked to the gene USH2 in chromosome 1. That characterization will allow to apply the genic therapy and both implants, mother cells and cochlear, as it corresponds.
Key words: family, Usher syndrome, hypoakusia, visual field, genetics, retinitis pigmentosa.
INTRODUCCIÓN
Usualmente, la retinosis pigmentaria se presenta aislada, pero en ocasiones puede concomitar con alguna otra enfermedad en la misma persona o familia, 1 como ocurre con el síndrome de Usher, que es un raro trastorno causado por una mutación genética, heredado de forma autosomal recesiva por su descendencia a través de los padres, lo cual significa que un niño (o niña) debe heredar un gen defectuoso de cada progenitor para padecer el mencionado síndrome y que si porta uno solo, será asintomático. 2-4
El síndrome de Usher (USH por sus siglas en inglés) es la manifestación clínica de una asociación de hipoacusia neurosensorial con retinosis pigmentaria (RP), que se inicia generalmente a los 10 años de edad, empeora lentamente con el tiempo y se caracteriza por mala visión nocturna, campo visual tubular e imágenes electrorretinogramáticas no registrables. 5-8
La hipoacusia neurosensorial impide que los nervios auditivos trasmitan entradas sensoriales al cerebro; pero en quienes padecen este síndrome, ello puede acompañarse de disfunción vestibular. 6
El síndrome de Usher ha sido considerado como la causa más frecuente de sordera-ceguera de origen genético en seres humanos y, de hecho, a su presencia se debe que más de 50 % de los neonatos nazcan sordociegos; además de ello, afecta aproximadamente a 10 % de los niños con hipoacusia profunda o severa. 9 La prevalencia de la disfunción varía entre 3,2-6,2 casos por cada 100 000 habitantes y en Cuba es la forma sindrómica más frecuente de la retinosis pigmentaria. 7
Desde el punto de vista clínico se diferencian 3 tipos, 10,11 partiendo principalmente del grado de hipoacusia y edad de inicio de la retinosis:
- Tipo I (USH1): Hipoacusia congénita severo-profunda, disfunción vestibular y aparición de la RP en la etapa prepuberal
- Tipo II (USH2): Hipoacusia congénita de carácter moderado-severo, función vestibular normal y comienzo de la RP en la segunda o tercera década de vida
- Tipo III (USH3): Hipoacusia poslingual progresiva, RP de inicio y gravedad variable, así como función vestibular afectada o no
En algunos pacientes, la variedad del cuadro sintomático obliga a clasificarlo como síndrome de Usher atípico. 12
Durante la investigación sobre retinosis pigmentaria en la provincia de Santiago de Cuba se encontró que en 3 hermanos se asociaba esa enfermedad con sordera; hallazgo que incentivó en las autoras el interés por realizar la caracterización oftalmológica, auditiva y genético-molecular de esa familia consanguínea.
MÉTODOS
Se efectuó un estudio de casos en el Centro Provincial de Retinosis Pigmentaria de Santiago de Cuba desde abril de 2010 hasta marzo de 2011, basado en una familia consanguínea compuesta por 25 miembros, distribuidos en 5 generaciones, entre los cuales 3 de sus componentes presentaban retinosis pigmentaria y sordera (2 varones y 1 fémina).
A tales efectos se indicaron los siguientes exámenes:
Oculares: agudeza visual, refracción, biomicroscopia, oftalmoscopia binocular, tonometría, campo visual con perimetría de Goldmann y estudios electrofisiológicos
Auditivos: otoscopia, audiometría, potenciales evocados auditivos y pruebas vestibulares
Genéticos: árbol genealógico y estudios moleculares
RESULTADOS
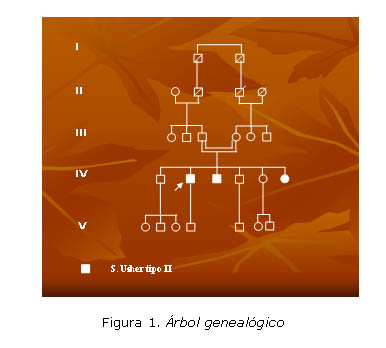
En el árbol genealógico (figura 1) se puso de manifiesto que 3 de los 25 integrantes de esta familia consanguínea estaban enfermos y compartían características familiares comunes (figura 2).
Los componentes IV2![]() IV3
IV3 ![]() y IV6
y IV6 ![]() tienen en común:
tienen en común:
- Comienzo en la etapa juvenil
- Mala visión nocturna
- Campos visuales reducidos
- Imágenes electrorretinogramáticas no registrables
- Hipoacusia
- Potenciales evocados auditivos de tallo cerebral: conducción no alterada en la vía auditiva, pero con retardo en el mecanismo de transducción periférica por hipoacusia neurosensorial
- Pruebas vestibulares con resultados normales
Características individuales
El paciente ![]() IV 2 se encontraba en el estadio III de la retinosis pigmentaria y presentaba campos visuales con reducción concéntrica a 10 grados en ambos en ambos ojos, a lo cual se adicionaban potenciales evocados visuales con latencia prolongada. La otoscopia del oído derecho mostró un timpano monomérico y ninguna alteración en el izquierdo, en tanto la audiometría reveló una grave hipoacusia neurosensorial bilateral.
IV 2 se encontraba en el estadio III de la retinosis pigmentaria y presentaba campos visuales con reducción concéntrica a 10 grados en ambos en ambos ojos, a lo cual se adicionaban potenciales evocados visuales con latencia prolongada. La otoscopia del oído derecho mostró un timpano monomérico y ninguna alteración en el izquierdo, en tanto la audiometría reveló una grave hipoacusia neurosensorial bilateral.
El paciente ![]() IV 3 se hallaba en el estadio II de la retinosis pigmentaria y tenía campos visuales con disminución concéntrica a 20 grados en ambos ojos, pero ningún resultado anormal de los potenciales evocados visuales en estos últimos. La otoscopia no evidenció alteraciones en los oídos, en contraste con la audiometría, que sí confirmó la existencia de una grave hipoacusia sensorial bilateral.
IV 3 se hallaba en el estadio II de la retinosis pigmentaria y tenía campos visuales con disminución concéntrica a 20 grados en ambos ojos, pero ningún resultado anormal de los potenciales evocados visuales en estos últimos. La otoscopia no evidenció alteraciones en los oídos, en contraste con la audiometría, que sí confirmó la existencia de una grave hipoacusia sensorial bilateral.
La paciente ![]() IV 6 clasificaba en el estadio II de la retinosis pigmentaria y la reducción concéntrica de su campo visual era de 25 grados, con resultados normales de los potenciales evocados visuales en ambos ojos y de la otoscopia en los 2 oídos; pero en cambio la audiometría puso de manifiesto una grave hipoacusia neurosensorial bilateral.
IV 6 clasificaba en el estadio II de la retinosis pigmentaria y la reducción concéntrica de su campo visual era de 25 grados, con resultados normales de los potenciales evocados visuales en ambos ojos y de la otoscopia en los 2 oídos; pero en cambio la audiometría puso de manifiesto una grave hipoacusia neurosensorial bilateral.
En otros familiares con riesgos se obtuvieron los siguientes resultados:
![]() III 3: electrorretinograma subnormal; otoscopia y audiometría normales
III 3: electrorretinograma subnormal; otoscopia y audiometría normales
![]() III 4: electrorretinograma, otoscopia y audiometría normales
III 4: electrorretinograma, otoscopia y audiometría normales
![]() IV 5: electrorretinograma, otoscopia y audiometría normales
IV 5: electrorretinograma, otoscopia y audiometría normales
Genética molecular
La electroforesis en gel de poliacrilamida, con estudio de ligamiento y marcadores microsatélites descritos para las regiones de los cromosomas 1 y 3, demostró la presencia del marcador D1S237, estrechamente ligado al gen USH2 en el cromosoma 1.
DISCUSIÓN
El síndrome de Usher en los miembros de la familia estudiada constituye la primera causa de sordera-ceguera hereditaria y es causado por múltiples mutaciones genéticas, algunas de las cuales ya identificadas y otras en vías de serlo. 13
Los 3 pacientes en cuestión, desde el punto de vista clínico, reunían todas las características distintivas del síndrome de Usher de tipo II, 14 pues el de tipo I incluye hipoacusia congénita severa, profunda disfunción vestibular y RP en la fase prepuberal, 15,16 en tanto el de tipo III se reconoce por hipoacusia poslingual progresiva, RP de inicio y gravedad variable, así como función vestibular afectada o no. 17 Las diferencias son notables entre una modalidad y otra.
De hecho, la interrelación del grupo multidisciplinario de retinosis pigmentaria de esta provincia con su homólogo del Centro Internacional, permitió confirmar el diagnóstico en los integrantes de la citada familia mediante la realización de estudios de genética molecular, que descartaron las otras variedades sindrómicas. 18
El conocimiento detallado y profundo de las causas genéticas de las hipoacusias ejerce un efecto inmediato sobre la implementación de terapias tendientes a garantizar la estimulación auditiva temprana en cualquiera de sus formas. 19,20 Actualmente pueden realizarse determinados estudios genéticos en laboratorios especializados del país, de suma importancia para la atención al paciente y la toma de decisiones, tanto personales como familiares.
Los avances tecnológicos y la experiencia adquirida sobre el funcionamiento de los mecanismos fisiológicos auditivo y ocular a lo largo de más de 50 años por la escuela cubana de retinosis pigmentaria, devienen una poderosa herramienta para el estudio y tratamiento del síndrome de Usher.
Finalmente, la caracterización de las distintas familias abrirá nuevos horizontes científicos y, por tanto, propiciará la aplicación de tratamientos más actualizados como la terapia génica y los implantes de células madres y cocleares, con lo cual se complementarán las aspiraciones de mejorar cada día más la salud y calidad de vida de la población cubana.
REFERENCIAS BIBLIOGRÁFICAS
1. Hernández Baguer R, Copello Noblet M, Dyce Gordon B, Rodríguez Alba M, Arce Álvarez A, Saint- Blancard Morgado G, et al. Retinosis pigmentaria: clínica, genética y epidemiología en estudio de familias habaneras. Rev Haban Cienc Méd 2008; 7(1). <http://www.ucmh.sld.cu/rhab/rhcm_vol_7num_1/rhcm17108.htm> [consulta: 7 febrero 2011].
2. Kimberling WJ, Hildebrand MS, Shearer AE, Jensen ML, Halder JA, Trzupek K, et al. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet Med 2010; 12(8):512-6.
3. Dalomon V, Elgoyhen AB. Hipoacusias de origen genético. Actualización. Rev Med Clin Condes 2009; 20(4):408-17.
4. Cortes Almela R, Cenjor Español C, Ayuso García C. Síndrome de Usher: aspectos clínicos, diagnósticos y terapéuticos <http://sid.usal.es/libros/discapacidad/17031/8-4-3/sindrome-de-usher-aspectos-clinicos-diagnosticos-y-terapeuticos.aspx>[consulta: 10 marzo 2011].
5. El síndrome de Usher. <http://spanish.hear-it.org/page.dsp?page=330> [consulta: 27 abril 2011].
6. Preda M, Damian C, Irimia A, Sollosy M, Ciuca CA, Totolin M. Usher síndrome. Oftalmologia 2008; 52(4):40-3.
7. Avances de la oftalmología cubana. <http://newsgroups.derkeiler.com/Archive/Soc/soc.culture.cuba/2005-12/msg00801.html>[consulta: 27 abril 2011].
8. Familiares y afectados. <http://www.bajavision.es/lateralfam.html> [consulta: 10 marzo 2011].
9. Causas genéticas. <http://ecodepadres.org/causas%20geneticas.html> [consulta: 20 junio 2011].
10. Síndrome de Usher. <http://www.sordoceguera.org/vc3/sordoceguera/sindrome_usher/sindrome_usher.php>[consulta: 20 junio 2011].
11. Síndrome de USHER. <http://www.foaps.es/la-sordoceguera/causas> [consulta: 27 abril 2011].
12. Soares Liarth JC, Atem Concalves E, Riveiro Concalves JO, Martins Neiva D, Macedo Leal FA. Síndrome de Usher: características clínicas. Arq Bras Oftalmol 2002; 65:457-61.
13. Halladas 32 mutaciones causantes del síndrome de Usher. <http://integracion.implantecoclear.org/index.php?option=com_content&view=article&id=267:halladas-32-mutaciones-causantes -del-usher&catid=1:latest-news&Itemid=18> [consulta: 27 abril 2011].
14. Hilgert N, Kahrizi K, Dieltjens N, Bazazzadegan N, Najmabadi H, Smith RJ, et al. A large deletion in GPR98 causes type IIC Usher syndrome in male and female members of an Iranian family. J Med Genet 2009; 46(4):272-6.
15. Pennings RJ, Kremer H, Deutman AF, Kimberling WJ, Cremers CW. From gene to disease; genetic causes of hearing loss and visual impairment sometimes accompanied by vestibular problems (Usher syndrome). Ned Tijdschr Geneeskd 2002; 146(49):2354-8.
16. Astuto LM, Weston MD, Carney CA, Hoover DM, Cremers CW, Wagenaar M, et al. Genetic heterogeneity of Usher syndrome: analysis of 151 families with Usher type I. Am J Hum Genet 2000; 67(6):1569-74.
17. Herrera W, Aleman TS, Cideciyan AV, Roman AJ, Banin E, Ben-Yosef T, et al. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Invest Ophthalmol Vis Sci 2008; 49(6):2651-60.
18. Tazetdinov AM, Dzhemileva LU, Khusnutdinova EK. Molecular genetics of Usher syndrome. Genetika 2008; 44(6):725-33.
19. Seeliger MW, Fischer MD, Pfister M. Usher syndrome: clinical features, diagnostic options, and therapeutic prospects. Ophthalmologe 2009; 106(6):505-11.
20. Blanchet C, Roux AF, Hamel C, Ben Salah S, Artières F, Faugère V, et al. Usher type I syndrome in children: genotype/phenotype correlation and cochlear implant benefits. Rev Laryngol Otol Rhinol (Bord) 2007; 128(3):137-43.
Recibido: 25 de junio de 2011
Aprobado: 15 de julio de 2011
MsC. Rásife Freyre Luque. Centro Provincial de Retinosis Pigmentaria, calle Centro Gallego No. 5, entre Aguilera y Anacaona, Ampliación de Terraza, Santiago de Cuba, Cuba.
Dirección electrónica:MsC.Rásife Freyre Luque

{kind=link}
{kind=link}