










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN vol.16 no.5 Santiago de Cuba mayo 2012
ARTÍCULO DE REVISIÓN
Bases moleculares de las enfermedades mitocondriales
Molecular basis of mitochondrial diseases
Dra. Aglais Arredondo Falagán, I Lic. Gleymis Venet Cadet,I Dra. Dra. Olivia Román Guerra I y MsC. Eglis Yanet Ramírez Delgado II
I Facultad de Medicina No. 2, Santiago de Cuba,Cuba.
II Hospital Infantil Norte "Dr. Juan de la Cruz Martínez Maceira", Santiago de Cuba, Cuba.
RESUMEN
Las mitocondrias son orgánulos subcelulares que tienen como misión principal la producción de energía, los cuales contienen su propio sistema genético que codifica un número pequeño de proteínas que forman parte del sistema de fosforilación oxidativa. En los últimos años han sido descubiertas mutaciones en el material genético mitocondrial que originan las enfermedades mitocondriales. Con el objetivo de contribuir a elevar y actualizar el conocimiento acerca del tema se realizó una revisión bibliográfica, donde se expone, además, la relación de las mitocondrias con afecciones como el alzheimer, el parkinson y la diabetes mellitus, por citar algunas.
Palabras clave: mitocondria, enfermedad mitocondrial, enfermedad neurodegenerativa, envejecimiento.
Abstract
Mitochondria are subcellular organelles whose primary role is to produce energy, which contain their own genetic system that encodes a small number of proteins that are part of the oxidative phosphorylation system. In recent years mutations in mitochondrial genetic material have been discovered, causing mitochondrial diseases. In order to increase and update the knowledge of the subject a literature survey was performed, where the relationship of mitochondria with conditions such as Alzheimer's, Parkinson's and diabetes mellitus, to name a few, is also stated.
Key words: mitochondria, mitochondrial disease, neurodegenerative disease, aging.
INTRODUCCIÓN
La mitocondria tuvo en el pasado un interés restringido para los estudiosos de la citología, la bioquímica metabólica y la bioenergética; sin embargo, desde hace unos años esta se encuentra en un primer plano de la actualidad de las ciencias biomédicas. Así, en la última década, muchas investigaciones se han dirigido hacia el estudio de esta organela, al ser considerada como el lugar donde convergen diferentes vías de señalización de muerte celular, tanto apoptóticas como necróticas.
Como bien se plantea, la mitocondria es una organela citoplasmática de características muy especiales. Su nombre proviene del griego mito (hilo) y chondros (cartílago). Por su origen endosimbiótico se convierte en un componente de gran importancia en la vida de la célula. 1,2
Todas sus características estructurales y funcionales evidencian que en el nacimiento de la célula eucariota, una bacteria fue fagocitada por un microorganismo de mayor tamaño con el fin de aprovecharse de la energía en forma de adenosín trifosfato (ATP) que esta fabricaba, mientras la célula anfitriona dotaba a su huésped de materias primas y protección. La simbiosis perfecta entre estos 2 organismos primitivos supone en uno de los pasos más importantes de la evolución. A lo largo del tiempo, estas bacterias fagocitadas fueron asumiendo tareas cada vez más relevantes dentro del funcionamiento interno de esa célula primitiva hasta convertirse en lo que hoy se conoce como mitocondria; esta hipótesis tiene entre sus fundamentos la evidencia de que las mitocondrias poseen su propio ADN. 1-3
Las mitocondrias son cilíndricas, aunque experimentan cambios de forma sutiles, derivados de su actividad. Normalmente se les representa en forma alargada. Su tamaño oscila entre 0,5 y 1 μm de diámetro y hasta 5 μm de longitud. Su número depende del tipo celular y de las necesidades energéticas de la célula. Una célula puede tener desde unas pocas mitocondrias hasta miles de ellas. El mayor número se encuentra en las células nerviosas, musculares y del hígado, por ejemplo: en los hepatocitos suelen hallarse entre 1 000 y 2 000 mitocondrias. Se encuentran ubicadas en las regiones de las células donde la demanda de energía es mayor, por lo que se desplazan de un lado a otro del citoplasma hacia las zonas necesitadas de energía. Los microtúbulos y sus proteínas asociadas intervienen en tales desplazamientos; no obstante, en algunos tipos celulares, como los espermatozoides, las células musculares y grasas, las mitocondrias permanecen en lugares fijos. Al conjunto de las mitocondrias de la célula se le denomina condrioma celular. 1, 4
Estas presentan una estructura con 2 compartimentos bien definidos (matriz y espacio intermembranal), delimitados por las membranas interna y externa, con características morfológicas, funcionales y de permeabilidad muy diferentes.
La membrana interna se caracteriza morfológicamente por presentar unas invaginaciones (crestas) con multitud de complejos enzimáticos y proteínas que regulan el paso de metabolitos. Esta resulta especialmente impermeable a iones, debido a su alto contenido en el fosfolípido cardiolipina y a la gran cantidad de bombas y transportadores específicos, como el translocador de nucleótidos de adenina. Por el contrario, la membrana externa carece de crestas y, en condiciones fisiológicas, su permeabilidad es menos selectiva, gracias a la presencia de una proteína denominada porina o canal aniónico dependiente de voltaje, que permite el paso de iones y metabolitos con pesos moleculares inferiores a 6 000 daltones. 1- 4
Las mitocondrias desempeñan diferentes funciones, una de las fundamentales es la respiración celular, pues mediante esta se obtiene energía metabolicamente utilizable por la célula, en ella tienen lugar algunas vías metabólicas como la cetogénesis, cetolisis, entre otras; también interviene en la remoción de Ca2+ del citosol, en la termogénesis y control de la apoptosis, son las principales generadores de especies reactivas del oxígeno en la célula y pueden provocar la muerte celular por necrosis en condiciones de estrés oxidativo. 5-7
Una de las particularidades de estos organelos es la de poseer un sistema genético propio con toda la maquinaria necesaria para su expresión, es decir, para replicar, transcribir y traducir la información genética que contiene.
El ácido desoxirribonucleico mitocondrial (ADNmt) humano es una molécula bicatenaria, circular, cerrada, sin extremos, con un peso molecular de 11 000 000 daltones aproximadamente, no está asociado con historias, se replica a partir de un solo punto de origen y es muy pequeño. En casi todos los tipos celulares la suma de los ADN tomados de todas las mitocondrias representa no más de 1 % del ADN nuclear. 8,9
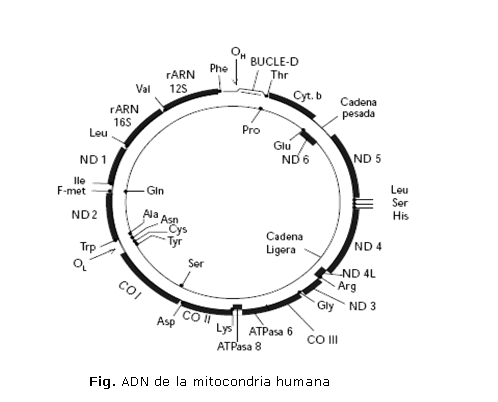
La secuencia del genoma mitocondrial del ser humano se conoce en su totalidad, no posee secciones no codificantes, está compuesto por 16 569 pares de bases y contiene un pequeño número de genes, distribuidos entre las cadenas H y L.
Cada mitocondria contiene entre 2 y 10 copias de la molécula de ADN. Frente a los 20 000 _ 25 000 genes del ADN cromosómico nuclear humano, el ADN mitocondrial tiene información para 37 genes: 2 ácidos ribonucleicos ribosómicos (rARN), componentes de los ribosomas específicos mitocondriales, 22 de transferencia (tARN), que son capaces de leer todo el código genético y 13 polipéptidos que forman parte de 4 de los 5 complejos multienzimáticos del sistema de fosforilación oxidativa (sistema Oxphos), etapa terminal de la ruta de producción de ATP. Estos péptidos corresponden a 7 subunidades del complejo I; una subunidad del complejo III; 3 subunidades del complejo IV y 2 subunidades del complejo V. El resto de los polipéptidos componentes de estos complejos, así como el complejo II completo, están codificados en el DNA nuclear.
La biogénesis de este sistema constituye un caso único en la célula, ya que para su formación se requiere la expresión coordinada de los 2 sistemas genéticos: el genoma mitocondrial, que solo aporta la información de un reducido número de proteínas y el del núcleo celular, que contiene el resto. 9-12
Otra característica importante del ADN mitocondrial es que no se recombina, lo cual implica que los únicos cambios que puedan haber ocurrido en el ADN mitocondrial se deben exclusivamente a mutaciones a lo largo de multitud de generaciones.
Presentan, además, una elevada tasa de mutación debido a 3 aspectos fundamentales: la generación de radicales de oxígeno por la cadena respiratoria, no posee histonas que protejan y presenta muy pocos sistemas de reparación. 12
En la figura se observa el ADN circular de la mitocondria humana en el que se representan sus 2 cadenas, con los genes correspondientes.
¿Por qué ha permanecido un ADN en la mitocondria?
No se tiene respuesta a esta pregunta, pero se han sugerido las causas siguientes:
- Las proteínas que están codificadas en el genoma mitocondrial presentan un alto grado de hidrofobicidad, lo cual dificultaría su importe a la mitocondria.
- Dichas proteínas podrían ser tóxicas en el citoplasma.
- La existencia de un genoma en la mitocondria permitiría una regulación local, rápida y más fina de la expresión de los genes que codifica.
- Es posible que todavía no haya terminado de transferir toda la información que contiene al núcleo, quizás por el código genético diferente que utiliza. 4
La genética del ácido desoxirribonucleico mitocondrial se diferencia de la del ADN nuclear por 4 aspectos fundamentales:
1. Herencia materna: El ADNmt se hereda por vía materna con un patrón vertical no mendeliano. La madre transmite su genoma mitocondrial a todos sus hijos, pero solamente las hijas lo pasarán a todos los miembros de la siguiente generación y así sucesivamente.
2. Poliplasmia: En cada célula hay cientos o miles de moléculas de ADNmt.
3. Segregación mitótica: Durante la división celular, las mitocondrias se distribuyen al azar entre las células hijas.
4. Alta velocidad de mutación: La tasa de mutación espontánea del ADNmt es 10 veces mayor que en el ADN nuclear.
¿PUDIERAN LAS MITOCONDRIAS ESTAR RELACIONADAS CON ALGUNAS ENFERMEDADES?
Las mutaciones en proteínas mitocondriales pueden conducir a un descenso en la producción de ATP, formación de radicales libres y alteraciones en la homeostasis del calcio en la célula, por ejemplo: en la paraplejía espástica hereditaria se encuentra mutada la paraplegina, una metaloproteasa mitocondrial que da lugar a defectos en la fosforilación oxidativa.
Otro ejemplo es la ataxia de Friedreich, en la que se encuentra mutada la proteína mitocondrial frataxina requerida para el mantenimiento de la homeostasis de hierro y el contenido en ADN. Las disfunciones mitocondriales también desempeñan una función esencial en otras enfermedades neurodegenerativas, tales como: Parkinson, Huntington y Alzheimer. Además, en sistemas donde no se lleva a cabo un "reemplazamiento" o "renovación" celular, como es el sistema nervioso, se pueden acumular mutaciones somáticas mitocondriales que, sumadas al descenso en la función mitocondrial, pueden ser la causa del envejecimiento y la senescencia. 13-15
Se considera que la mitocondria es la fuente generadora de especies reactivas del oxígeno más importante. El incremento en la formación de O2- y H2O2 se justifica con el hallazgo de que en el envejecimiento se modifican las condiciones del flujo de electrones en la cadena de transporte de estos. 16
El genoma mitocondrial es muy susceptible al ataque por radicales libres producido en la propia mitocondria, los que reaccionan con el ADN mitocondrial, lo lesionan y dan lugar a deleciones y mutaciones. Esto produce cambios con el tiempo, se compromete la formación de ATP y la síntesis de proteínas. Al respecto, existen múltiples evidencias que corroboran la importancia de la disfunción mitocondrial en la patogénesis de la destrucción celular que causa envejecimiento; el estrés oxidativo es el principal inductor de esas alteraciones. 17, 18
La actividad de las reacciones enzimáticas que abastecen energéticamente a la célula, puede verse reducida o comprometida debido a un aporte inadecuado de oxígeno a las células, como ocurre en procesos de aterosclerosis, anemia o alcoholismo. Una reducción de su actividad se ha observado en pacientes con enfermedad de Parkinson. En este sentido, algunas neurotoxinas o sus metabolitos pueden ser causa o contribuir a dicha enfermedad, inhibidores de estos complejos enzimáticos y que hoy día constituyen parte de las herramientas farmacológicas utilizadas como inductores en modelos experimentales para el estudio de los mecanismos celulares que tienen lugar en estas enfermedades degenerativas. 19-21
Diferentes estudios han sugerido que mutaciones del ADNmt y disfunciones de la cadena respiratoria pueden estar implicadas en la patogénesis de la diabetes mellitus.
En primer lugar, mutaciones del ADNmt asociadas con encefalopatías mitocondriales se han identificado bien en estos pacientes; en segundo lugar, es más frecuente heredar la diabetes mellitus de una madre afecta que de un padre con la enfermedad, lo cual sugiere una herencia materna de los factores predisponentes. En estudios in vitro se ha demostrado que son necesarios el DNAmt y la cadena respiratoria intacta para la liberación de insulina mediada por la glucosa. Este hecho sugiere que mutaciones del DNAmt u otras causas que alteren la función de la fosforilación oxidativa de las células beta de los islotes pancreáticos, pueden originar una reducción de la secreción de insulina y, por tanto, desarrollar la diabetes mellitus. 22
ENFERMEDADES MITOCONDRIALES
Los caracteres moleculares básicos y peculiares del sistema genético mitocondrial se descubrieron al inicio de los años 80 y en 1988 se encontraron las primeras mutaciones asociadas a enfermedades. Desde entonces, el número de mutaciones en el ADNmt y de enfermedades asociadas se ha incrementado significativamente, pues han sido encontradas más de 150 mutaciones (más de 100 deleciones y unas 50 mutaciones puntuales).
Se designa con el nombre de enfermedades mitocondriales a un grupo de trastornos cuya característica común es la deficiencia en la síntesis de ATP. El término de citopatías mitocondriales se reserva para disfunciones de la cadena respiratoria mitocondrial. 23-26
Los primeros datos epidemiológicos de las enfermedades del ADNmt se centraron en la población blanca de Europa del Norte, residente en el noreste de Inglaterra y fueron enunciados por el grupo del doctor Turnbull, en Newcastle, Reino Unido, quienes demostraron que los defectos en el ADNmt son la causa de enfermedad en 6,57 de cada 100 000 individuos de la población adulta trabajadora y que 7,59 por cada 100 000 adultos y niños no afectados corren el riesgo de desarrollar una de estas enfermedades. En total, 12,48 por 100 000 individuos (1 de cada 8 000) tienen o presentan un riesgo de padecer una enfermedad causada por daños en el ADNmt. 11
De hecho, las manifestaciones clínicas son muy heterogéneas, pueden presentarse como enfermedades fatales en el recién nacido, en los primeros años de vida, durante la adolescencia y la adultez o como enfermedades degenerativas. Presentan múltiples síntomas y signos por afectación de diversos tejidos y órganos no relacionados desde los puntos de vista fisiológico y embriológico, fundamentalmente: corazón, cerebro, músculo esquelético y en particular la musculatura ocular por tener grandes demandas de energía oxidativa. Entre las manifestaciones clínicas más comunes se encuentran una o varias de las siguientes: desórdenes motores, accidentes cerebrovasculares, convulsiones, demencia, intolerancia al ejercicio, ptosis, oftalmoplejía, retinopatía pigmentaria, atrofia óptica, ceguera, sordera, cardiomiopatía, disfunciones hepática y pancreática, diabetes mellitus, defectos de crecimiento, anemia sideroblástica, seudoobstrucción intestinal, nefropatías, acidosis metabólica, entre otras. 9, 27, 28
La heterogeneidad de las manifestaciones clínicas, morfológicas y bioquímicas de las enfermedades del ADNmt hace que su clasificación se base muy frecuentemente en las características genéticas de las mutaciones, a pesar de que en algunos casos, una misma mutación puede dar lugar a fenotipos clínicos muy diversos. Así, dichas enfermedades se pueden dividir en 3 grandes grupos, según estén asociadas a mutaciones puntuales, a reorganizaciones o a disminución de número de copias del ADNmt. Al respecto, en la severidad de la manifestación de la enfermedad intervienen varios factores: la naturaleza de la mutación, el grado de heteroplasmia, los requerimientos energéticos del tejido y la capacidad de este para compensar el daño celular. A continuación se presenta un resumen de las enfermedades más comunes asociadas a estos tipos de mutaciones. 12, 23
1. ENFERMEDADES ASOCIADAS CON MUTACIONES PUNTUALES
Dado el alto índice de mutación del ADNmt, como se ha indicado anteriormente, es posible encontrar un gran número de mutaciones puntuales; sin embargo, la mayoría son silenciosas, pues no causan ningún tipo de defecto. Las patológicas se pueden encontrar tanto en los genes de tARN, como en los codificantes de proteínas, y responden siempre a un tipo de herencia materna, 29 entre estas se encuentran:
A. Síndrome MERRF, por sus siglas en inglés: Epilepsia mioclónica y fibras rojas rasgadas.
B. Síndrome MELAS, por sus siglas en inglés: Encefalomiopatía mitocondrial, acidosis láctica y episodios parecidos a un accidente vascular encefálico.
C. Síndrome NARP: Se presenta con neuropatía, ataxia y retinosis pigmentaria.
D. Neuropatía óptica hereditaria de Leber
2. ENFERMEDADES ATRIBUIBLES A REORGANIZACIONES DEL ADNmt POR INSERCIONES O DELECIONES, O AMBAS
Además de las mutaciones puntuales, el ADNmt puede sufrir otro tipo de daños, tales como: pérdida de parte de este (deleciones) o la adición de un nuevo fragmento del ADN (duplicaciones), que, como en los casos anteriores, afectan a la biogénesis del sistema Oxphos y, por tanto, a la síntesis de ATP.
Hoy día hay descritos más de 100 tipos de deleciones y solo unos cuantos casos de inserciones. Este tipo de mutaciones suelen ser espontáneas, probablemente causadas por daños en genes nucleares que controlan la replicación del ADNmt, aunque han sido notificados casos de herencia materna. 30, 31
Los 3 síndromes más comunes en los que se presentan deleciones son: el de Pearson, la oftalmoplejía progresiva externa crónica y el de Kearns-Sayre, los cuales se caracterizan por una deleción larga o, más raramente, por una duplicación de 1,3 a 7,6 kb. 32,33
3. ENFERMEDADES ASOCIADAS A DEPLECIONES DE ADN MITOCONDRIAL
El tercer tipo de daños en el genoma mitocondrial que puede causar enfermedades no se debe a mutaciones propiamente dichas, sino a una disminución de los niveles del ADNmt. El espectro clínico que produce la depleción es muy variado. Los casos descritos hasta ahora afectan fundamentalmente a niños con combinaciones variables de miopatía, nefropatía o hepatopatía, miopatía infantil fatal por dificultad respiratoria y algún otro con implicación multisistémica. La depleción puede estar producida por mutaciones en genes nucleares que controlan el número de copias del ADNmt. Por tanto, es un trastorno de herencia mendeliana que afecta a la coordinación núcleo-mitocondria y que parece ser autosómico recesivo. 34, 35
Debido al doble origen genético nuclear y mitocondrial del sistema Oxphos, las enfermedades genéticas mitocondriales, como ya se ha analizado, pueden estar originadas por las mutaciones en genes del ADNmt con herencia materna, así como también por mutaciones en genes nucleares que codifican proteínas mitocondriales y ocasionan un mal funcionamiento de procesos que se desarrollan en las mitocondrias, como alteraciones de enzimas, ARN, componentes de la cadena de transporte de electrones y sistemas de transporte de la membrana interna; muchas de ellas afectan al músculo esquelético y al sistema nervioso central. Por lo tanto, en la clasificación de las enfermedades mitocondriales desde el punto de vista genético deben tenerse en cuenta las alteraciones del ADN nuclear: 23
1. Alteraciones de los genes que codifican proteínas mitocondriales: Mutaciones en genes para subunidades de la cadena respiratoria mitocondrial (complejos I y II) y mutaciones en proteínas ancilares (complejos III, IV y V)
2. Alteraciones en la importación de proteínas mitocondriales
3. Alteraciones en la comunicación intergenómica: Deleciones múltiples del ADNmt, depleción de este y defectos en su traducción
4. Alteraciones en el medio lipídico: Síndrome de Barth
5. Alteraciones en la motilidad/fusión/fisión mitocondrial: Atrofia óptica y paraplejía espástica familiar
¿Cuándo sospechar que hay una disfunción mitocondrial? No hay una característica única para identificar una enfermedad mitocondrial. Los pacientes presentan varios problemas que pueden surgir desde el nacimiento hasta la edad adulta madura. 36
Piense en las mitocondrias cuando exista una "enfermedad común" con características atípicas que la distingan del resto, cuando hayan 3 o más órganos involucrados, cuando se presenten recaídas recurrentes o cuando aparezcan brotes de infección en una enfermedad crónica.
El diagnóstico de estas afecciones resulta bastante difícil por la gran variedad de manifestaciones clínicas que presentan, así como también por la complejidad de los exámenes complementarios que se realizan, entre los cuales figuran: la biopsia de músculo y el análisis molecular para la búsqueda de mutaciones en el ADNmt. Hasta el momento no existe un tratamiento que las cure, de manera que la conducta médica debe estar dirigida a mejorar la calidad de vida de los pacientes.
Tras años de investigación, el Ministerio Británico de Sanidad está dispuesto a autorizar un nuevo tratamiento con el objetivo de "borrar" en el laboratorio una herencia genética defectuosa y evitar el nacimiento de niños con enfermedades mitocondriales. Por primera vez, se utilizaría material genético de 3 personas (óvulos de 2 mujeres y el esperma de un varón) para lograr el nacimiento de un bebé sano.
Después de una fecundación in vitro, se extraen los núcleos del espermatozoide del progenitor y el óvulo de la madre, que contienen genes de los padres y se dejan atrás las mitocondrias defectuosas. Los núcleos se implantan posteriormente en el óvulo de una mujer sana al que se le ha despojado su núcleo y conserva sus mitocondrias sanas. Estas últimas no llevan información genética que defina las características de una persona, de modo que los bebés que nazcan por este procedimiento se parecerán a sus padres legales y no padecerán de enfermedades mitocondriales.
CONCLUSIONES
Las mitocondrias no solo intervienen en la producción de energía metabólicamente utilizable por la célula, ellas van más allá. El hecho de que posean su propio ADN hace, que al producirse un daño de este, se originen afecciones causadas por defectos en el sistema de fosforilación oxidativa, las enfermedades mitocondriales. Las mitocondrias también se encuentran relacionadas con la génesis de algunas enfermedades como el Alzheimer, el Parkinson y la diabetes mellitus, entre otras, por lo que es de gran importancia continuar su estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Campos Y. Panorámica de actualidad sobre las enfermedades mitocondriales. Madrid: Centro de Investigación Hospital 12 de Octubre; 2007.
2. Ruiz L. La mitocondria: mucho más que una fábrica de energía [citado: 16 May 2011]. Disponible en: http://www.ecogenesis.com.ar/index.php?sec=articulo.php&Codigo=47
3. Morrison J. Genetics of mitochondrial diseases. European J Human Genetics. 2004;12: 686_687.
4. Hernández Rivas J. Bases moleculares de las enfermedades mitocondriales. Salamanca:Universidad Autónoma; 2008.
5. AD therapeutic approaches tap complement, mitochondrial antioxidant [citado: 16 May 2011]. Disponible en: http://www.alzforum.org/new/detail.asp?id=2213
6. Bell EL, Klimova T, Eisenbart J, Moraes CT, Murphy MP, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007; 177: 945.
7. Selak M, Duran R, Gottllieb E. Redox stress is not essential for the pseudo-hypoxic phenotype of succinate dehydrogenase deficient cells. Biochim Biophys Act. 2006; 1757:567-72.
8. Finsterer J, Harbo HF, Baets J, Van Broeckhoven C, Di Donato S, Fontaine B, et al. EFNS guidelines on the molecular diagnosis of mitochondrial disorders. Eur J Neurol. 2009;16(12):1255-64.
9. Rubio González T, Verdecia Jarque M. Las enfermedades mitocondriales: un reto para las ciencias médicas. MEDISAN 2004 [citado: 16 May 2011]; 8(1). Disponible en:http://bvs.sld.cu/revistas/san/vol8_n1_04/san08104.htm
10. Gazdíková K, František G. Mitochondrial nephrology [citado: 16 May 2011]. Disponible en: http://www.springerlink.com/content/x15hr807555229w0/fulltext.pdf?MUD=MP
11. Solano A, Playán A, López Pérez MJ, Montoya J. Enfermedades genéticas del ADN mitocondrial humano. Salud Pública Mex. 2001; 43:151-61.
12. Tucker EJ, Compton AG, Thorburn DR. Recent advances in the genetics of mitochondrial encephalopathies. Curr Neurol Neurosci Rep. 2010;10(4):277-285
13. Madamanchi R, Runge M. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007; 100(4): 460-73.
14. Majamaa K, Winqvist S. A 3-year clinical follow-up of adult patients with 3243A>G in mitochondrial DNA. Neurology. 2006; 66(10): 1470-5.
15. Yao J, Irwin R, Zhao L, Nilsen J, Hamilton R, Diaz Brinton R, et al. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci. 2009;106(34):14670-5.
16. Kienhöfer J, Häussler DJ, Ruckelshausen F, Muessig E, Weber K, Pimentel D, et al. Association of mitochondrial antioxidant enzymes with mitochondrial DNA as integral nucleoid constituents. FASEB J. 2009; 23(7):2034-44.
17. Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling Cardiovasc Res. 2009; 81(3):449-56.
18. Fedotcheva, N, Sokolov A, Kondrashova N. Nonenzymatic formation of succinate in mitochondria under oxidative stress. Free Radic Biol Med. 2006; 41:56-64.
19. Luoma P, Melberg A, Rinne J. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004; 364(9437):875-82.
20. Davidzon G, Greene P, Mancuso M, Klos KJ, Ahlskog JE, Hirano M, et al. Early-onset familial parkinsonism due to POLG mutations. Ann Neurol. 2006; 59:859-62.
21. Hudson G, Schaefer A, Taylor RW, Tiangyou W, Gibson A, Venables G, et al. Mutation of the linker-region of POLG1 associated with PEO with parkinsonism. Arch Neurol. 2007; 64:553-7.
22. Quintana L, Sanjurjo P. Alteraciones de la b-oxidación y del sistema de carnitina. En: Sanjurjo P, Baldellou A. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Editorial Ergón; 2001.p. 275-94.
23. Andreu L, Gonzalo Sanz R. Las enfermedades mitocondriales: una clasificación para el siglo XXI. Neurol. 2004; 19(1):15-22.
24. Salvatore D, Michelangelo M. Mitochondrial diseases: therapeutic approaches. Biosci Rep. 2007; 27: 125-37.
25. Saneto DO, Nicole IW, Niklas D, Bruce H. Suspected mitochondrial disorder. Inherited Metabolic Diseases. 2010; 13(1):325-33.
26. Scharfe C, Horng-Shing Lu H, Neuenburg JK, Allen RA, Guan-Cheng L, Klopstock T, et al. PLoS Comput Biol. 2009; 5(4): e1000374.
27. Finsterer J. Hematological manifestations of primary mitochondrial disorders. Acta Haematol. 2007; 118 (2): 88-98.
28. Nava Reyes HJ, Zamudio Cortés P, Quiroz Cabañas Y, Martínez Ramírez I, Espinosa Pérez A, García Cruz A, et al. La disfunción mitocondrial como posible causa de la falla orgánica múltiple asociada a la sepsis severa. Rev Inst Nal Enf Resp Mex. 2009;22(1): 37-47.
29. Gómez S, Castro O, Benavent P. MELAS: claves del diagnóstico y tratamiento en la Unidad de Cuidados Intensivos. Med Intensiva. 2008; 32(3):147-50.
30. Vedolin L, Fischinger C. Conventional MRI and MR spectroscopy in nonclassical mitochondrial disease: report of three patients with mitochondrial DNA deletion. Child's Nervous System. 2006; 22(10):1355-9.
31. Kam Ming Au, Shing Chi Lau. Mitochondrial DNA deletion in a girl with Fanconi's syndrome. Pediatric Nephrology. 2007; 22(1): 136-40.
32. Martín Hernández E, García Silva MT, Quijada Fraile P, Martínez de Aragón A, Cabello A, Martín MA. Síndromes de Pearson y de Kearns-Sayre: dos enfermedades mitocondriales multisistémicas, debidas a deleciones en el ADN mitocondrial. Acta Pediatr Española. 2010; 68(9): 451-9.
33. Martín Hernández E, García Silva MT, Vara J, Campos Y, Cabello A, Muley R, et al. Renal pathology in children with mitochondrial diseases. Pediatr Nephrol. 2005; 20(9): 1299-1305.
34. Mitochondrial Medicine Society's Committee on Diagnosis, Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008; 94(1):16-37.
35. Debray F, Lambert M, Chevalier I, Robitaille Y, Decarie JC, Shoubridge EA, et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics. 2007; 119 (4):722-33.
36. Di Mauro S, Hirano M. Pathogenesis and treatment of mitochondrial disorders. Adv Exp Med Biol. 2009; 652:139-170.
Recibido: 1 de abril de 2011.
Aprobado: 20 de marzo de 2012.
Aglais Arredondo Falagán. Facultad de Medicina No. 2, avenida Cebreco, km 1 ½, reparto Pastorita, Santiago de Cuba, Cuba. Correo electronico:aglais@medired.scu.sld.cu

{kind=link}