










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN vol.17 no.9 Santiago de Cuba set. 2013
ARTÍCULO ORIGINAL
Diagnóstico por pesquisa neonatal de metabolopatías congénitas en el Centro Provincial de Genética Médica de Santiago de Cuba
Diagnosis of inborn errors of metabolism by neonatal screening at the Provincial Center of Medical Genetics of Santiago de Cuba
Dr. Daniel Fernando Rojas Bernal, MsC. Tatiana Góngora Wilson, MsC. Hilda Gertrudis Álvarez Valiente, MsC. Gloria Seisdedos Gómez y Dra. Aimé Macías Quintosa
Hospital Infantil Sur, Santiago de Cuba, Cuba.
RESUMEN
Se realizó un estudio descriptivo y transversal de 20 niños de 0 a 5 años de edad con metabolopatías congénitas (fenilcetonuria, galactosemia, deficiencia de biotinidasa, hiperplasia suprarrenal congénita e hipotiroidismo congénito), quienes habían sido diagnosticados a través de la pesquisa neonatal, procedentes de todos los municipios de Santiago de Cuba, y fueron atendidos en el Centro Provincial de Genética Médica desde el 2006 hasta el 2011, a fin de caracterizarles según algunas variables clínicas y epidemiológicas. En la provincia de Santiago de Cuba se obtuvo una baja tasa de incidencia de los trastornos metabólicos congénitos detectados en la pesquisa neonatal, con una mayor frecuencia del hipotiroidismo congénito (55,0 %). De igual manera, los pacientes mostraban escasas manifestaciones clínicas, las cuales, además, eran leves. Los resultados de la serie reflejaron la presencia de un diagnóstico y tratamiento oportunos, unidos a una adecuada atención pediátrica.
Palabras clave: metabolopatías congénitas, hipotiroidismo congénito, niños, pesquisa neonatal, centros de genética.
ABSTRACT
A descriptive and cross sectional study was conducted in 20 children from 0 to 5 years of age with inborn errors of metabolism (phenylketonuria, galactosemia, biotinidase deficiency, congenital adrenal hyperplasia and congenital hypothyroidism), coming from all the municipalities of Santiago de Cuba, who were diagnosed through neonatal screening and attended in the Provincial Center of Medical Genetics from 2006 to 2011, in order to characterize them according to some clinical and epidemiological variables. In Santiago de Cuba province a low rate of incidence of congenital metabolic disorders diagnosed in neonatal screening was obtained, with a higher frequency of congenital hypothyroidism (55.0%). Similarly, patients had a few clinical manifestations, which also were mild. The results of the series revealed the presence of an early diagnosis and treatment, with adequate pediatric care.
Key words: inborn errors of metabolism, congenital hypothyroidism, children, neonatal screening, genetics centers.
INTRODUCCIÓN
Las metabolopatías congénitas, los trastornos metabólicos o las enfermedades metabólicas hereditarias se deben a alteraciones bioquímicas que modifican la estructura o función de las moléculas proteicas que intervienen en el metabolismo. El defecto estructural se expresa en la actividad de enzimas u otros compuestos que intervienen en los procesos de transporte, síntesis o degradación de sustancias químicas, y bloquean el flujo de metabolitos o de otros productos.1
En Cuba comenzó a implementarse masivamente el diagnóstico de la fenilcetonuria a partir del año 1986 mediante el Programa Cubano de Diagnóstico Manejo y Prevención de Enfermedades Genéticas y Defectos Congénitos -- incluido en el Programa de Tecnología Avanzada del Ministerio de Salud Pública--, que contiene la pesquisa de fenilcetonuria, hipotiroidismo congénito, galactosemia, deficiencia de biotinidasa e hiperplasia suprarrenal congénita, con el objetivo de establecer un diagnóstico precoz y aplicar un tratamiento oportuno para evitar daños irreparables.
Igualmente, en este país la incidencia de la fenilcetonuria es de 1 por cada 45-50 000 nacidos vivos, mucho menor que en los países del norte de Europa, donde la incidencia es de 1 por 10 000 nacidos vivos o quizás más. Por su parte, en EE.UU. y Canadá se notifica una incidencia de 1 por 15 000 o 20 000, y en México de 1 por 25 000.2
Respecto al hipotiroidismo congénito, en el mundo se registra una frecuencia de 1 por 100 000 para el hipotiroidismo terciario y secundario, 1 de cada 4 000 nacidos vivos para el primario, mientras que para el hipotiroidismo transitorio se refiere 1 por cada 200 000-50 000.3,4 En Cuba la cifra es de 1 por 2 503 nacimientos con vida.2
Teniendo en cuenta la inexistencia de estudios previos en la provincia de Santiago de Cuba sobre aspectos clínicos y epidemiológicos de las metabolopatías congénitas, los cuales son de gran importancia para el seguimiento y pronóstico de estos pacientes, se consideró realizar esta investigación, para caracterizar los afectados detectados mediante la pesquisa neonatal.
MÉTODOS
Se realizó un estudio descriptivo y transversal de 20 niños de 0 a 5 años de edad con metabolopatías congénitas (fenilcetonuria, galactosemia, deficiencia de biotinidasa, hiperplasia suprarrenal congénita e hipotiroidismo congénito), quienes habían sido diagnosticados a través de la pesquisa neonatal, procedentes de todos los municipios de Santiago de Cuba, y fueron atendidos en el Centro Provincial de Genética Médica desde el 2006 hasta el 2011, a fin de caracterizarles según las siguientes variables clínicas y epidemiológicas: años del diagnóstico, edad al momento del estudio, sexo, tipo de metabolopatía congénita, evaluación nutricional, manifestaciones clínicas de cada enfermedad y evolución clínica.
La pesquisa de los trastornos metabólicos congénitos estuvo en correspondencia con la secuencia de trabajo para establecer el diagnóstico clínico-bioquímico-enzimático de dichas afecciones, a través de la aplicación de las técnicas de pesquisa masiva del sistema ultramicroanalítico (SUMA) y de otros métodos en el Centro Nacional de Genética Médica, previa coordinación con este.
RESULTADOS
El hipotiroidismo congénito resultó la metabolopatía congénita más frecuente (tabla 1), con 55,0 % del total, seguida en orden descendente de la hiperplasia suprarrenal congénita, con 5 pacientes, para 25,0 %. A partir del año 2009, de forma general se evidenció un incremento en el número de afectados; igualmente, en el 2009 se diagnosticó el mayor número de niños con estos trastornos (6 de ellos, para 30,0 %).
La tabla 2 muestra el predominio del sexo masculino (razón de 1,5:1), fundamentalmente en aquellos con hiperplasia suprarrenal congénita e hipotiroidismo congénito, con 60,0 y 63,6 % de varones, respectivamente.
Respecto a la incidencia de las metabolopatías congénitas (tabla 3), se obtuvo una baja incidencia de cada uno de estos trastornos, con tasas de valores inferiores a 1,5 por cada 10 000 nacidos vivos. Se observó mayor incidencia en los pacientes con hipotiroidismo congénito, con una tasa de 1,3 y una razón de 1:7 232, en tanto la menor incidencia fue de 0,25 para la fenilcetonuria y la deficiencia de biotinidasa, con una razón de 1:39 777.
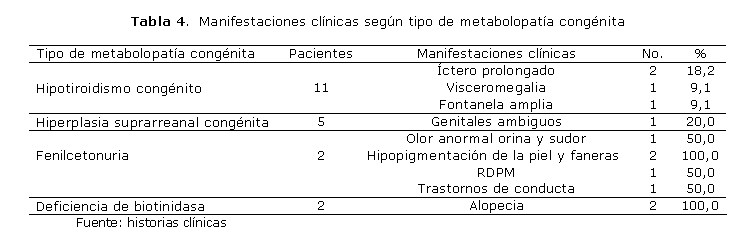
En cuanto a la incidencia de los síntomas y signos más frecuentes (tabla 4), se halló que todos los afectados con fenilcetonuria presentaban hipopigmentación de la piel y faneras, mientras que 50,0 % de ellos manifestaban olor anormal del sudor y la orina y retardo del desarrollo psicomotor (RDPM). En los pacientes con hiperplasia suprarrenal congénita, solo 20,0 % mostró ambigüedad sexual, mientras que en los niños con hipotiroidismo congénito primó el íctero prolongado (18,2 %). Los 2 afectados con deficiencia de biotinidasa mostraron alopecia como única manifestación clínica.
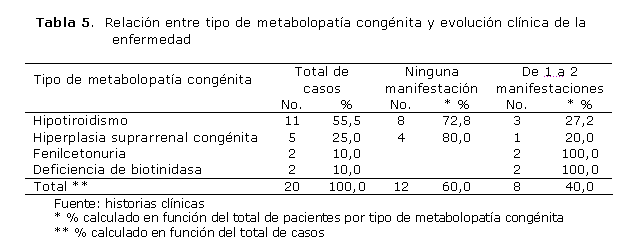
Al relacionar las metabolopatías congénitas con el desarrollo clínico de cada enfermedad (tabla 5), se obtuvo un total de 12 afectados (60,0 %) sin ningún tipo de manifestación; de ellos, 8 padecían hipotiroidismo congénito, para 40,0 %. Ninguno de los pacientes presentó más de 2 manifestaciones clínicas.
En relación con la evaluación nutricional, todos los niños mantuvieron un buen crecimiento pondoestatural.
DISCUSIÓN
En Cuba hay una total cobertura del Programa Cubano de Pesquisa Neonatal de Enfermedades Metabólicas Hereditarias, al igual que el seguimiento y tratamiento posterior para los casos que se diagnostican como positivos. En el año 2007 se inició en Santiago de Cuba la aplicación del SUMA, con el uso de la técnica de ultramicroELISA en la pesquisa neonatal de fenilcetonuria e hipotiroidismo congénito, que antes se investigaban por métodos menos sensibles, y se incorporó al Programa el estudio de la deficiencia de biotinidasa, la hiperplasia suprarrenal congénita y la galactosemia, lo que explica el porqué a partir de esta fecha se incrementó el número de diagnósticos de metabolopatías congénitas.
Según han planteado otros autores,3-6 el hipotiroidismo congénito constituye la enfermedad endocrino-metabólica más frecuente, la cual causa retardo mental prevenible. El argumento anterior concuerda con los resultados de la serie, que mostraron mayor frecuencia de la enfermedad.
Algunos investigadores2,7 coinciden en que no existe predominio de un sexo sobre otro entre los que padecen hipotiroidismo congénito. Sin embargo, un estudio7 realizado en México notifica un mayor número de varones con hipotiroidismo, lo cual se corresponde con lo obtenido en esta casuística, pero difiere de lo referido en otra serie8 que mostró primacía de las féminas.
Respecto a la fenilcetonuria y la deficiencia de biotinidasa no existe supremacía en alguno de los sexos.7-9
Conforme a los datos expuestos en el Anuario Estadístico cubano del 2011, en Santiago de Cuba ocurrieron más nacimientos de varones, por lo que la preponderancia del sexo masculino en este estudio pudiera estar condicionada por tal situación.
Los hallazgos en los pacientes con hipotiroidismo congénito de la casuística ocurrieron en edades tempranas. Al respecto, en la bibliografía médica se refiere la aparición de signos incluso durante la vida prenatal, por lo que no debe extrañar la presencia de visceromegalia y fontanela anterior amplia desde los primeros días de vida extrauterina de estos pacientes, lo que también justifica el desarrollo de los diferentes programas neonatales. Precisamente esto ha hecho posible que no se encuentren manifestaciones avanzadas de la enfermedad, como retardo del desarrollo psicomotor y pondoestatural, daño óseo, entre otras características propias de un hipotiroidismo congénito no tratado.5,6
Por otra parte, la hiperplasia suprarrenal congénita como consecuencia de la deficiencia de la enzima 21 hidroxilasa se reconoce como la causa más común de genitales ambiguos.2,3
De la misma manera, la alopecia como único síntoma en todos los afectados con deficiencia de biotinidasa, pudiera deberse a la ingestión irregular de vitamina H o biotina, que es un suplemento nutricional. El diagnóstico temprano de la enfermedad y el tratamiento con dosis substitutivas de biotina, previenen la aparición de las manifestaciones clínicas y de las alteraciones bioquímicas.10
La presencia de hipopigmentación de la piel y el pelo en los pacientes con fenilcetonuria, coincide con los resultados expuestos en otros trabajos sobre el tema.3,11,12
Al analizar la evolución clínica según el tipo el tipo de metabolopatía congénita, se observaron pocas manifestaciones clínicas el período que duró este estudio, desde que se realizaran los diagnósticos en la pesquisa del 2006 hasta que finalizó la investigación en el 2011. Con estos resultados se demostró una evolución favorable de los niños, lo que estuvo en correspondencia con un diagnóstico adecuado, un tratamiento efectivo y oportuno, buena calidad del Programa y correctos seguimiento y control de todos los pacientes.
En cuanto a la evaluación nutricional de los niños, todos presentaban el peso y la talla adecuados; resultados que demuestran el logro del Programa activo de pesquisa y del seguimiento sistemático de estos pacientes, al impedir la evolución de síntomas incapacitantes. Contrariamente a lo expresado, otros autores observaron retraso o aceleración del desarrollo pondoestatural.2,3,5,6
Para dar por concluido, la provincia de Santiago de Cuba exhibió una baja tasa de incidencia de las metabolopatías congénitas diagnosticadas por medio de la pesquisa neonatal, con una mayor frecuencia del hipotiroidismo congénito. La aparición de pocas manifestaciones clínicas, síntomas leves y un buen crecimiento pondoestatural constituyó expresión del diagnóstico y tratamiento oportunos en los niños afectados, así como de la adecuada atención pediátrica.
REFERENCIAS BIBLIOGRÁFICAS
1. Vela Amieva M, Jiménez Sánchez G, Cicerón Arellano I, Velásquez Arellano A. Guía para el diagnóstico de los errores innatos del metabolismo. México, DF.: Academia Mexicana de Pediatría; 1998.
2. Borbolla Vacher L, García Martínez DA. Genética médica. En: De la Torre Montejo E, Pelayo González-Posada EJ. Pediatría. T 1. La Habana: Editorial Ciencias Médicas; 2006. p. 277-310.
3. Lantiga Cruz A. Enfermedades genéticas y defectos congénitos en la atención primaria. En: Álvarez Sintes R, Hernández Cabrera G, Báster Moro JC, García Núñez RD, Louro Bernal I, Céspedes Lantigua LA, et al. Medicina general integral. Salud y medicina. 2 ed. La Habana: Editorial Ciencias Médicas; 2008.
4. Cuba. Ministerio de Salud Pública. Dirección Nacional de Registros Médicos y Estadísticas de Salud. Anuario Estadístico de Salud 2012. La Habana: MINSAP [citado 26 Ago 2012]; 2013. Disponible en: http://files.sld.cu/dne/files/2013/04/anuario2012.pdf
5. Vela Amieva M, Gamboa Cardiel S, Pérez Andrade ME, Ortiz Cortés J, González Contreras CR, Ortega Velázquez V. Epidemiología del hipotiroidismo congénito en México. Salud Pública Méx. 2004; 46(2): 141-8.
6. Vela Amieva M, Belmont Martínez L, Fernández Lainez C, Ramírez Frías C, Ibarra González I. Frecuencia de enfermedades metabólicas congénitas susceptibles de ser identificadas por el tamiz neonatal. Acta Pediatr Mex. 2009; 30(3): 156-62.
7. Restrepo Carlos M. Genética. En: Fundamentos de pediatría. 3 ed. Medellín: Printer Colombiana; 2006; t IV. p. 333-55.
8. Repetto G, Durán G. Genética y enfermedades metabólicas. En: Guiraldes E, Ventura Juncá P. Manual de pediatría [citado 26 Ago 2012]. Disponible en:http://escuela.med.puc.cl/paginas/publicaciones/ manualped/GeneticaEnfMetab.html
9. Alvear Sedan CC, De Cortina B, Arrieta H, Alayón A. Tamizaje para hipotiroidismo congénito en Cartagena. Rev Pediatría [citado 5 Sep 2012]. Disponible en:http://www.encolombia.com/medicina/pediatria /pedi36101-tamizaje1
10. González Reyes EC, Marrero González N. Deficiencia de Biotinidasa. Bioquimia (México). 2002 [citado 12 Sep 2012]; 27(003). Disponible en:http://www.medigraphic.com/pdfs/bioquimia/bq-2002/bq023d .pdf
11. Therrel BL. Advances in neonatal screening: Proceedings of the 6th International Neonatal Screening Symposium, Austin, Texas, 16-19 November 1986 and the 5th National Neonatal Screening Symposium, Austin, Texas, 20 November 1986. Ámsterdam: Excerpta Medica; 1987. p. 35-9.
12. Fenilcetonuria [citado 12 Sep 2012]. Disponible en:http://salud.discapnet.es/Castellano/Salud/Discapacidades/Discapacidades%20Mentales/Fenilcetonuria/Paginas/Fenilcetonuria.aspx
Recibido: 12 de abril de 2013.
Aprobado: 23 de mayo de 2013.
Daniel Fernando Rojas Bernal. Hospital Infantil Sur, avenida "24 de Febrero", nr 402, Santiago de Cuba, Cuba. Correo electrónico:tatygw@medired.scu.sld.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}