










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Mayer-Rokitansky-Küster-Hauser (MRKHS por sus siglas en inglés) es una anomalía congénita causada por graves defectos en el desarrollo de los conductos de Müller, que se reconoce por una amenorrea primaria en pacientes fenotípicamente femeninas, quienes a pesar de carecer de los dos tercios superiores de la vagina o de esta completa, además del útero y las trompas, asociados a malformaciones renales, tienen ovarios funcionantes.1,2
Se trata de una rara enfermedad que afecta a 1 de cada 5 000 mujeres, cuyo síntoma principal es la ausencia de la menarquía o primera regla del ciclo menstrual, de origen desconocido, pero caracterizada por la convergencia de múltiples factores, entre los cuales no se desecha una probable causa genética, la falta de receptores de las hormonas sexuales en los conductos de Müller, así como un déficit de la enzima galactosa 1 fosfato uridiltransferasa.3,4
La asistencia a una paciente joven con esa afección tan infrecuente, atendida en un hospital materno ubicado en el municipio de Santiago de Cuba, justificó la preparación del presente artículo en la modalidad de caso clínico, con vistas a incrementar el número de trabajos científicos publicados sobre el tema en Cuba y otras partes del mundo, así como el interés que sigue despertando su descripción.
Caso clínico
Se describe el caso de una paciente de 21 años de edad, fenotípicamente normal a simple vista, que acudió en 2017 a la consulta de ginecología del Hospital Materno Norte “Tamara Bunke Bider” de Santiago de Cuba no solo por presentar amenorrea, sino por el hecho de que al tratar de tener relaciones sexuales, estas fueron muy difíciles y dolorosas, de manera que no llegaron a consumarse, pues comúnmente la vagina se encuentra acortada.
Antecedentes patológicos personales y familiares: ninguno, según lo referido por ella en la entrevista médica
Antecedente obstétricos materno: nacida de un parto eutócico a término
Examen físico
Examen ginecológico
Labios menores poco desarrollados y clítoris normal
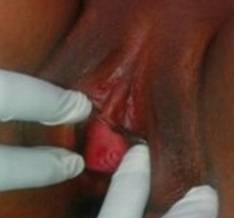
Introito vaginal: ausencia de la membrana del himen, con el sitio ocupado por un rafe medio extendido en dirección vertical (Fig. 1).
Exámenes complementarios
Cariotipo 46XX
Ecografías de pelvis y riñones: resultados normales
Resonancia magnética de pelvis: en el corte sagital, ausencia de útero y vagina (Fig. 2).

Resonancia magnética de ovarios derecho e izquierdo: con ovulación (Fig.3).
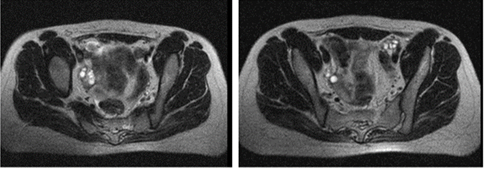
Fig. 3 En las imágenes de la resonancia magnética se aprecian los ovarios derecho e izquierdo con ovulación.
Diagnóstico definitivo: síndrome de Mayer-Rokitansky-Küster-Hauser.
Comentarios
El trastorno se conoce como síndrome de Mayer-Rokitansky-Küster-Hauser en honor a las contribuciones de August Franz Joseph Karl Mayer, quien describió en 1829 el primer caso de ausencia de vagina en una recién nacida; Carl Freiherr von Rokitansky, quien publicó en 1838 los resultados de las autopsias de 19 adultas con agenesia uterovaginal; HermannKüster, quien observó las anomalías renales urológicas en 1810, así como Georges André Hauser, quien en 1861 identificó en estas pacientes, determinadas malformaciones esqueléticas asociadas.3,4
Barrecheguren et al5 plantean que existen 2 tipos de síndromes: el típico y el atípico; en el primero, la amenorrea primaria presenta alteraciones solamente en las estructuras müllerianas, mientras que en el segundo se acompañan de agenesias renales congénitas y esqueléticas.
Por su parte, si bien unos autores6 señalan que los cariotipos y fenotipos de las pacientes con este cuadro clínico son normales, otros7,8 opinan que no se ha logrado establecer una asociación causal del trastorno con algunas anomalías generales o patrón de herencia genética y que actualmente se incluyen el grupo de las alteraciones del desarrollo sexual.
También se apunta que la causa exacta de este síndrome se desconoce y que en la mayoría de los casos se diagnostica en mujeres sin antecedentes familiares de esa condición; sin embargo, aunque históricamente se ha valorado la posibilidad de que pudiera ser consecuencia de una enfermedad materna o de la exposición fetal a sustancias nocivas contenidas en ciertos medicamentos, hasta ahora no se ha encontrado asociación entre el síndrome y la medicación u otro tipo de factor de riesgo, así como tampoco se ha confirmado la influencia de algún gen en la aparición de la anomalía, a pesar de conjeturarse que los factores genéticos y ambientales podrían tener participación en el proceso a través de una herencia autosómica dominante.9,10
El diagnóstico clínico suele establecerse antes de los 20 años, basado en la amenorrea primaria y la incapacidad de penetración vaginal; pero no debe obviarse el diferencial, pues también las pacientes con síndrome de insensibilidad androgénica presentan acortado el conducto de la vagina.3
Lógicamente, la ausencia de útero impide que las mujeres con este síndrome puedan quedar embarazadas; pero sería posible que lograsen su descendencia genética por medio de la fertilización in vitro.

