










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las mucopolisacaridosis (MPS) constituyen un grupo heterogéneo de trastornos hereditarios causados por deficiencia de una enzima lisosómica, necesaria para catabolizar carbohidratos complejos, conocidos como glucosaminoglicanos (GAG). Son enfermedades multisistémicas, crónicas y progresivas de baja frecuencia. La acumulación de GAG puede causar daño progresivo a una gran variedad de tejidos y provocar rigidez articular, malformaciones esqueléticas, retraso en el crecimiento, déficit pulmonar, así como anomalías oculares, hepáticas, cardíacas y neurológicas. Todas las enfermedades son de herencia autosómica recesiva, salvo la MPS II que es ligada al cromosoma X. La incidencia global se estima en 1/22 500, según las series de casos estudiados. Existen 7 tipos de MPS (MPS I, II, III, IV, VI, VII y IX); algunas de ellas con varios subtipos. En total son 11 las enfermedades que se diferencian por el defecto enzimático, por el tipo de GAG eliminado en orina y por el fenotipo clínico.1,2
A pesar de que las enfermedades de almacenamiento lisosomal (EAL) se encuentran entre las primeras afectaciones genéticas para las que se elucidaron los defectos bioquímicos primarios, aún queda mucho por conocer sobre los mecanismos patogénicos subyacentes. Los GAG por sí mismos no son tóxicos, pero el exceso puede producir alteraciones en la estructura celular de diferentes tejidos, principalmente en hueso, cartílago y córnea; asimismo, las consecuencias celulares de la acumulación de estos sustratos están determinadas por el tipo y grado de material que se almacena, el tipo de células donde se acumulan dichos sustratos sin degradar y las consecuencias directas o indirectas que tiene el almacenamiento lisosómico en procesos celulares básicos como el tráfico intracelular y la autofagia.3) Ciertamente, es un desafío diferenciar el grado en que cada una de estas alteraciones contribuye cuantitativamente a la patogénesis. Por lo tanto, la elucidación de los mecanismos fisiopatológicos de estas entidades clínicas es una tarea compleja y exigente, que requiere de un enfoque integrado que va desde la genética molecular hasta la bioquímica, la biología celular y la inmunología.
A lo anterior se adiciona que otros defectos celulares y de desarrollo tempranos pueden estar contribuyendo a los fenotipos clínicos de los pacientes con EAL. En este sentido se plantea que las afectaciones del lisosoma conducen a una capacidad reducida de estos para fusionarse con los autofagosomas y los endosomas, lo que trae consigo el bloqueo de los procesos de degradación celular y de autofagia. Este hecho está considerado en uno de los modelos descritos para explicar la patogénesis de las EAL. Por consiguiente, varios de los sustratos que son degradados por estas vías, como los agregados de proteínas poliubiquitinadas y las mitocondrias disfuncionales, se acumulan y originan la muerte celular.4) Por otra parte, las deficiencias de las enzimas lisosomales y las afectaciones en estas vías de degradación pueden causar disfunción mitocondrial, lo cual trae consigo la producción excesiva de especies reactivas del oxígeno (ERO) y la desregulación de la homeostasis del calcio, lo que sumado a una reducción de las defensas antioxidantes, puede conducir a respuestas de muerte celular por apoptosis.5
Teniendo en cuenta que en Cuba aún no existen antecedentes documentados sobre los niveles de marcadores de daño oxidativo y defensa antioxidante en pacientes con MPS, y basados en lo anteriormente expuesto, los autores se propusieron realizar la presente investigación para evaluar los parámetros bioquímicos y de estrés oxidativo en dichos pacientes.
Métodos
Se realizó un estudio observacional, analítico, de casos y controles de 7 pacientes con diagnóstico clínico y bioquímico de MPS: MPS I (n=2), MPS II (n=2), MPS III (n=1) y MPS IV (n=2), con edades comprendidas entre 1-12 años. El grupo control estuvo conformado por 21 niños aparentemente sanos, con igual rango de edad. Para la selección de este grupo se comprobó previamente el estado de salud mediante anamnesis, examen físico y exámenes de laboratorio clínico (pruebas de función hepática, glucemia, creatinina, lipidograma, hemoglobina y leucograma) para comprobar la presencia o no de otras enfermedades que pudieran relacionarse con el estrés oxidativo. Como se trata de una enfermedad de origen genético se tuvo en cuenta que estos niños no presentaran antecedentes familiares de MPS.
En ambos grupos se verificó que en el momento del estudio los niños no estuvieran consumiendo suplementos vitamínicos o antioxidantes.
La investigación se desarrolló en los laboratorios de Genética Bioquímica y Estrés Oxidativo del Centro Nacional de Genética Médica. Las muestras de todos los participantes fueron remitidas desde las consultas de genética clínica de los hospitales pediátricos Pedro Borrás, de Centro Habana, y William Soler, en el período 2016-2017.
Como muestra biológica se utilizó sangre venosa. La extracción se realizó en los laboratorios de los referidos centros asistenciales, en condiciones de ayuna de 12 horas. El suero, el plasma y el lisado de glóbulos rojos se obtuvieron según lo establecido internacionalmente para tales fines.
Para cada una de las determinaciones se tuvieron en cuenta los criterios internos de control de la calidad. Las concentraciones de ácido úrico, creatinina, hierro, ferritina, transferrina, ceruloplasmina y calcio, así como las actividades de las transaminasas se determinaron en suero, mediante un analizador químico Chemical Analyser Elecsys de 2010.
En el plasma se determinaron las concentraciones de malondialdehído (MDA) y los productos avanzados de oxidación de proteínas (PAOP), como marcadores de daño oxidativo, así como los niveles de grupos tioles libres (R-SH). En los lisados de eritrocitos se evaluaron las actividades enzimáticas de la superóxido dismutasa citosólica (SOD1), la catalasa (CAT) y la glutatión peroxidasa (GPx) como los marcadores de capacidad de defensa antioxidante. Todas las técnicas de marcadores de estrés oxidativo (EO) se realizaron mediante métodos espectrofotométricos descritos previamente por el grupo de trabajo.6
Los resultados se expresaron como medias ± desviación estándar (DE). Se compararon las medias aritméticas de cada una de las variables de respuesta para ambos grupos (casos y controles), mediante la prueba U de Mann-Whitney para muestras independientes. Como criterio de significación se tomó el valor de p<0,05. Para el análisis estadístico se utilizó el programa SPSS, versión 13.0 para Windows.
Por cuestiones éticas, todos los participantes se incluyeron en el estudio luego de que sus representantes legales o tutores emitieran voluntariamente su consentimiento, según los principios éticos de la Declaración de Helsinki de 1975, modificada en el 2013. El protocolo de la investigación se expuso a la aprobación del Comité de Ética de las investigaciones del Centro Nacional de Genética Médica.
Resultados
De los 7 pacientes incluidos en el estudio, 71,0 % correspondieron al sexo masculino y la edad promedio fue de 6 años. Los resultados de los estudios hemoquímicos se describen en la tabla 1.

En relación con las variables de daño oxidativo (tabla 2), se evidenció un incremento en las concentraciones plasmáticas de MDA y PAOP entre los pacientes con mucopolisacaridosis y los controles.
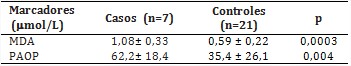
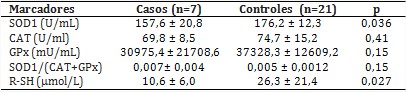
La actividad intraeritrocitaria de la SOD1 disminuyó considerablemente en el grupo con MPS en comparación con los controles. Las actividades de la CAT y de la GPx no mostraron diferencias entre los grupos estudiados; a pesar de ello, los pacientes tienden a tener disminuida las actividades de estas enzimas antioxidantes.
Al evaluar la eficiencia de este sistema antioxidante, determinado a través de la relación SOD1/GPx+CAT (tabla 3), se apreció un ligero incremento de esta proporción en los pacientes, pero no difiere con la obtenida en los niños aparentemente sanos. Por otra parte, se observaron diferencias en las concentraciones plasmáticas de los grupos tioles libres entre los grupos de estudio.
Discusión
La deficiencia de enzimas lisosómicas y las afectaciones en vías como la autofagia y la endosomal se reconocen como los defectos primarios en las EAL; sin embargo, evidencias recientes señalan la función que desempeña la disfunción mitocondrial en el inicio y la progresión de estas enfermedades. Por su parte, la pérdida de la homeostasis mitocondrial trae consigo la producción excesiva de ERO y la desregulación en el metabolismo del calcio, dos eventos claves que están interconectados.5)
En las EAL, a consecuencia de los defectos primarios en el metabolismo lisosómico, se acumulan los GAG, metabolitos tóxicos que pueden generar un ambiente prooxidante. Datos derivados de estudios en modelos animales de MPS han confirmado que la acumulación intralisosomal de estos productos provocan directa o indirectamente el incremento en la producción ERO, mediado fundamentalmente por eventos inflamatorios y alteraciones en la homeostasis mitocondrial.5
Los resultados obtenidos en la presente investigación indican que los pacientes con estas MPS presentan un aumento significativo en las concentraciones plasmáticas del MDA, producto final de la peroxidación lipídica (PL). El aumento de este proceso oxidativo puede traer como consecuencia cambios en la permeabilidad y pérdida de la fluidez de las membranas celulares.7 En correspondencia con estos hallazgos, estudios similares refieren el incremento a nivel sistémico de la PL en pacientes con MPS I y II y en aquellos con la enfermedad de Fabry;8,9 resultados que confirman que la PL es un proceso que tiene lugar en los pacientes con EAL.
Los productos avanzados de oxidación de proteínas se producen como resultado de la acción de agentes oxidantes como el ácido hipocloroso, producido por la acción de la mieloperoxidasa, como parte de las respuestas inflamatorias. Se sugiere que estas proteínas modificadas, generadas durante la inflamación, activan los fagocitos mononucleares que actúan sobre estas células como mediadores proinflamatorios de manera similar a las citocinas.10
En consecuencia, los PAOP además de ser un buen marcador de daño oxidativo a las proteínas plasmáticas, constituyen un indicador de la ocurrencia de procesos inflamatorios. En el presente estudio, las diferencias encontradas para este parámetro entre pacientes y controles, sustentan la función de los procesos inflamatorios en la patogénesis de las MPS, descrito por otros autores,11,12) así como el incremento en las afectaciones oxidativas de las proteínas detectadas en dichos pacientes.7,9,11,13) En la literatura científica no se hallaron referencias en cuanto a la concentración de estos productos de oxidación en afectados con MPS, por lo que este constituye el primer informe al respecto.
La SOD1 es una de las primeras líneas de defensa antioxidante intracelular, encargada de transformar el anión superóxido en H2O2. Posteriormente, la CAT y la GPx lo descomponen, por lo que forman parte primordial de la respuesta antioxidante del eritrocito.14) Los pacientes con MPS muestran una disminución en la actividad de la SOD1, sin cambios en las actividades de la CAT y la GPx con respecto a los controles. Estas alteraciones se traducen en una tendencia al desequilibrio en la relación entre estas 3 enzimas metabolizadoras de ERO (SOD1/CAT+GPx), lo que implicaría que los pacientes presenten deficiencias en estos sistemas antioxidantes y se generen altos niveles de H2O2 y, por tanto, aumento en los procesos de PL,14) tal y como se observa en el presente estudio. Por otra parte, la actividad de la SOD1 puede verse afectada negativamente por el incremento de procesos de inflamación, así como en las deficiencias nutricionales de cobre.15)
De acuerdo con los resultados de esta serie, los autores consideran que la inflamación podría explicar la baja actividad de la SOD1 descrita, dado los niveles normales encontrados en la ceruloplasmina, indicador empleado para evaluar el metabolismo del cobre. Acorde a estos resultados, en un estudio realizado en pacientes con MPS IIIB se apreció el incremento del daño oxidativo con una baja expresión de la SOD1;9 sin embargo, en otras investigaciones no se describen alteraciones en las actividades de estas enzimas antioxidantes.11,13
La determinación de los tioles proteicos libres permite cuantificar los grupos sulfhidrilos referidos, fundamentalmente, a los residuos de cisteínas libres y al glutatión reducido (GSH) presentes en una muestra desproteinizada. Este compuesto es muy estudiado en las enfermedades relacionadas con el EO, ya que es uno de los más sensibles a las perturbaciones oxidativas. Además, los niveles de GSH en sangre reflejan su contenido en tejidos menos accesible y de esta manera es un indicador del estado redox.16,17)
En la casuística, resultados mostraron una disminución en el contenido de grupos tioles libres en el grupo de los casos, lo que pudiera estar relacionado con el aumento de su requerimiento a nivel intracelular en reacciones oxidativas.18) En concordancia con estos hallazgos, en un estudio efectuado en pacientes con MPS del tipo IVA, se obtuvo que el contenido de GSH estaba muy disminuido y esto se correlacionó negativamente con el daño oxidativo a las proteínas.11 En su conjunto, los resultados en los marcadores de EO indican la presencia de condiciones oxidativas a nivel sistémico, con alteraciones en la capacidad de respuesta antioxidante en los pacientes con las MPS estudiadas.
La alteración en los niveles intracelulares de calcio (Ca2+) se señala como una de las afectaciones que comúnmente aparece en las EAL.19 En el presente estudio, los pacientes con MPS mostraron un incremento del calcio extracelular, sin alteraciones en el resto de los marcadores hemoquímicos medidos, lo que sugiere un aumento en la liberación de este ión al plasma. En estudios recientes se refiere que las afectaciones en la homeostasis del Ca2+ afectan funcionalmente a los lisosomas y a las mitocondrias, pero también actúan sobre cascadas de señalización de muerte celular, por mecanismos que pueden diferir entre las diferentes EAL.19,20
De manera general, los resultados derivados de la presente investigación sugieren que entre los factores comunes en las MPS analizadas figuran la presencia de daño oxidativo, la reducción de las defensas antioxidantes y los niveles elevados de Ca a nivel sistémico. Estos eventos, unidos a los defectos primarios en el metabolismo lisosomal, influyen en los mecanismos patogénicos de estos trastornos.
Finalmente, los hallazgos derivados de esta investigación podrían ser el soporte para estudios posteriores, en los que se puede analizar la aplicación de estrategias terapéuticas basadas en la utilización de antioxidantes y el empleo de estos parámetros para realizar el seguimiento de estos pacientes.

