










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El coronavirus del síndrome respiratorio agudo grave de tipo 2: SARS-CoV-2 (siglas del inglés severe acute respiratory syndrome coronavirus 2) ha ocasionado una pandemia mundial, la COVID-19 (coronavirus disease-2019), con millones de personas infectadas y fallecidas, además de incalculables pérdidas económicas y sociales.1,2,3)
Con vistas a controlar la COVID-19, se han indicado medidas de aislamiento e higienización, así como la administración de diferentes medicamentos y vacunas; sin embargo, la aparición de nuevas variantes del SARS-CoV-2, debido a mutaciones propias de los virus, ha alarmado a la comunidad científica internacional, por la posibilidad de que estas evadan los mecanismos de inmunidad natural y adquirida.4
Tal situación podría condicionar negativamente la erradicación de la pandemia y convertir la infección del coronavirus en más contagiosa y peligrosa, con el consecuente incremento de la morbilidad y mortalidad, lo que ya ha sido demostrado en algunas variantes genéticas del SARS-CoV-2 de rápida propagación mundial, a pesar de los aspectos controversiales al respecto.5
Las mutaciones son sucesos frecuentes en el ciclo de vida de los virus; con el incremento de las replicaciones, es probable que aparezcan variantes peligrosas que escapen del sistema inmunitario, sobre todo en regiones con alta densidad poblacional y malos controles de la pandemia por sistemas sanitarios deficientes o mala gestión gubernamental.6,7
Una nueva variante del SARS-CoV-2 presenta mutaciones que la diferencian de la variante predominante que circula en la población. Dichas variaciones genéticas son importantes, pues suelen afectar la estructura y las funciones de las proteínas virales, lo que puede incidir en la capacidad de transmisión de los virus, en la susceptibilidad a los tratamientos y las vacunas, y puede generar una mayor virulencia o cambios en la patogenia.8,9
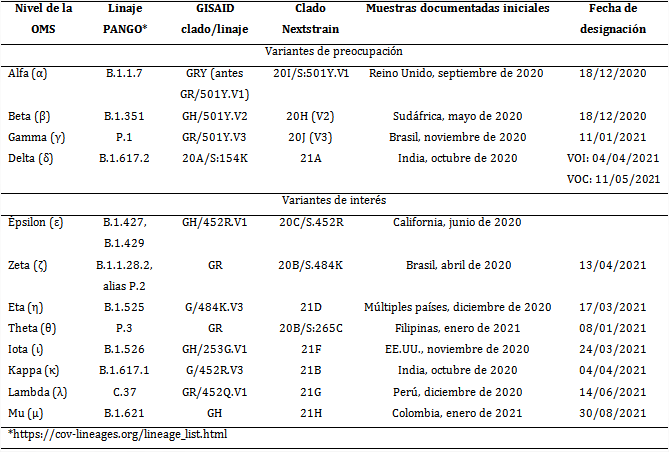
Las variantes del SARS-CoV-2 se clasifican en variantes preocupantes (VOC, por sus siglas del inglés variant of concern) y variantes de interés (VOI, siglas del inglés variant of interest).10 Las VOC descritas son alfa, beta, gamma y delta, mientras que entre las VOI se mencionan épsilon, zeta, eta, theta, iota, kappa, lambda y mu. Todas son investigadas intensamente en el orbe.11,12,13,14
En esta revisión bibliográfica se describen las características de estas nuevas variantes genéticas del SARS-CoV-2, con énfasis en su probable impacto clínico. Se aclara que existe mucha bibliografía sobre el tema y que es probable que aparezcan nuevas variantes o que las VOI se conviertan en VOC como fenómeno natural en la evolución de los virus.
Desarrollo
Mutaciones
El genoma del SARS-CoV-2 acumula una tasa de mutaciones estimada en aproximadamente 10−6 ―10−4 por ciclo replicativo. Ese es el origen de los “mutantes”, que pueden extinguirse si se genera una desventaja adaptativa, o que pueden prosperar si se proveen ventajas adaptativas. Así se originan las “variantes de escape del sistema inmunitario”.6,7
A medida que el virus experimenta variaciones genéticas, también puede alterar su manifestación, impactando sobre la transmisibilidad, la gravedad del estado clínico, el diagnóstico de laboratorio, el tratamiento, la eficiencia de las vacunas y las medidas de control. Por ende, resulta importante identificar las alteraciones genómicas que se producen en el SARS-CoV-2.15,16,17,18
El método de referencia para la identificación de las variantes genéticas es la secuenciación del genoma completo, aunque se han desarrollado técnicas de reacción en cadena de la polimerasa (PCR) en tiempo real para la identificación de mutaciones.7 Esto ha permitido que la secuenciación de genomas del SARS-CoV-2 esté disponible para la comunidad científica.
Las mutaciones más importantes del SARS-CoV-2 son las que alteran la proteína S, que tiene una función clave en la entrada del virus a las células. Cuando se incrementa la replicación de los virus, es más probable que aparezcan mutaciones en sus genomas, las cuales pueden alterar la susceptibilidad a la respuesta inmunitaria, la malignidad de la infección y/o la capacidad de transmisión del virus.7,16
Aunque los términos “mutante”, “variante” y “cepa” del virus son usados indistintamente en las descripciones epidemiológicas del SARS-CoV-2, no significan lo mismo. Las mutaciones son cambios en la secuencia de nucleótidos del genoma y los virus que las portan son llamados “mutantes”.6 No obstante, en esta revisión se tratarán estos términos como sinónimos.
SARS-CoV-2
El SARS-CoV-2 es un virus de la familia Coronaviridae, específicamente del género Betacoronavirus, cuyo genoma contiene una cadena simple de ácido ribonucleico (ARN) de casi 30 000 nucleótidos y genes codificantes de proteínas estructurales y no estructurales. Está organizado en el siguiente orden del extremo 5’ al 3’: marco de lectura abierto (open reading frame, ORF) 1ab (réplicas) y las proteínas S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N y ORF10.1,5,19,20
Dos tercios del ARN viral codifican dos grandes y solapadas ORF nombradas ORF1a y ORF1b, o ORF1ab juntas, que producen proteínas no estructurales (nsps). ORF1ab es una gran poliproteína (∼21,291 nucleótidos) que codifica 16 proteínas no estructurales: proteína líder, nsp2, nsp3, nsp4, proteinasa semejante a 3C, nsp6, nsp7, nsp8, nsp9, nsp10, ARN polimerasa dependiente de ácido desoxirribonucleico (RdRp) o Nsp12, helicase, 3’-5’ exonucleasa, endo-ARNsa, 2’-O-ribosa metilltransferasa y nsp11.5,21
La RdRp es el componente clave que regula la síntesis de ARN viral con la asistencia de Nsp7 y Nsp8. Además, 5 proteínas accesorias son codificadas por los genes ORF3a, ORF6, ORF7a, ORF8 y ORF10.20
Como se muestra en la figura 1 -tomada de Desimmie et al22-, el SARS-CoV-2 tiene una apariencia esférica, con proyecciones externas que le confieren un aspecto de corona; contiene cuatro proteínas estructurales (S, E, M y N) y dieciséis proteínas no estructurales (nsp1-16). La proteína de la membrana (M) le confiere forma y soporte a la partícula del virus e interactúa tanto con la S como con la nucleoproteína (N), que recubre el ARN viral, y la proteína de la envoltura (E), que impulsa el ensamblaje del virión y la formación de brotes y que tiene una actividad de canal iónico relacionada con la patogénesis viral.1,15,22,23
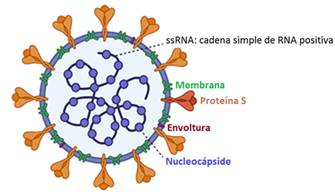
La proteína S (del inglés spike) interviene en la entrada del virus a la célula hospedera al unirse a la enzima convertidora de angiotensina 2 (ACE2) y dirigir la fusión de la membrana viral con la membrana hospedera. Esta fusión depende del clivaje de la proteína S por proteasas de la célula hospedera, lo cual ocurre después de la unión de los viriones a la membrana celular o durante la maduración y salida de los viriones.23,24,25,26
La importancia de la proteína S en la infección del SARS-CoV-2 la convierten en diana terapéutica para fármacos y vacunas anti-COVID-19.27,28,29,30 Al respecto, seguidamente se ofrecen más detalles.
Proteína S
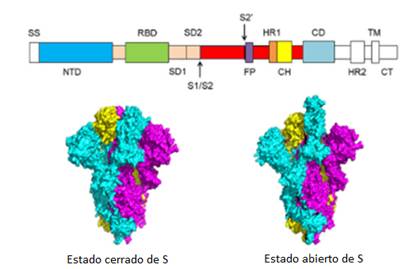
Los coronavirus entran a las células huésped mediante la proteína S, que es transmembranal, formada por trímeros (tres subunidades de 1273 aminoácidos) que protruyen en la superficie viral y dos subunidades funcionales S1 y S2.1,13
La subunidad S1 contiene un dominio N terminal (NTD) y un dominio de unión al receptor (RBD). La función de la subunidad S1 es unirse al receptor ACE2 sobre la célula diana mediante RBD.1
La subunidad S2 contiene el péptido de fusión (FP, siglas del inglés fusion peptide), la repetición heptapeptídica 1 (HR1, heptad repeat 1), la hélix central (CH, central helix), el dominio conector (CD, connector domain), la repetición heptapeptídica 2 (HR1, heptad repeat 2), el dominio transmembranal (TM, transmembrane domain) y la cola citoplasmática (CT, cytoplasmic tail).1
La función de la subunidad S2 es fusionar las membranas de los virus y las células diana.1 El sitio de escisión entre las subunidades S1 y S2 se llama sitio de clivaje o de escisión de proteasa S1/S2 (S1/S2 protease cleavage site).
Para todos los coronavirus las proteasas del huésped escinden la proteína S en el sitio de clivaje S2/S1 para activar las proteínas, lo cual es crítico para fusionar las membranas de los virus y las células del huésped. La figura 2, publicada por Wang et al,1 representa a la proteína S.
Nomenclatura
Pese a que habitualmente, tras la aparición de mutaciones, los términos cepa y variante se emplean como sinónimos, en sentido puro se debería considerar la inducción de cambios en el virus o no. No obstante, en este texto dichos términos se usarán indistintamente.2,7
Existen dos sistemas para la vigilancia epidemiológica: Phylogenetic Assignment of Named Global Outbreak (PANGO) y Nextstrain. El sistema de linaje PANGO posee un prefijo alfabético y un sufijo que contiene hasta tres números separados por puntos que indican sublinajes. Sin embargo, como el sistema solo permite tres niveles jerárquicos, la introducción de un nuevo sufijo puede dificultar la identificación del linaje ancestral. Además, el linaje de un virus no siempre se corresponde con sus mutaciones, ya que este puede presentar alteraciones adicionales relevantes sin que se le asigne un nuevo linaje.7,13
En la nomenclatura de las diferentes variantes del SARS-CoV-2 con frecuencia se hace referencia al país o lugar de detección (por ejemplo: cepa británica). Este sistema múltiple de clasificación usualmente genera confusiones. Por ello, la Organización Mundial de la Salud (OMS) adoptó un sistema más simple sustentado en el uso de letras griegas para denominar a las diversas variantes.7
Las mutaciones con cambios en los aminoácidos se denominan con una clave alfanumérica que inicia con una letra mayúscula para indicar el código del aminoácido que ha sido sustituido, luego va un número que señala la posición del cambio y, finalmente, otra letra mayúscula correspondiente al código del nuevo aminoácido. Así, por ejemplo, la mutación N501Y indica que el aminoácido asparagina (N) ha sido sustituido por una tirosina (Y) en la posición 501.2,7
Cepa ‘salvaje’ u original
A continuación se describirán las principales variantes del SARS-CoV-2, informadas hasta agosto de 2021.
La cepa original que inició la pandemia en diciembre 2019 portaba la mutación 484E en la proteína S, llamada cepa Wuhan-Hu-1. Entre enero y febrero de 2020 emergió una variante con una sustitución D614G [sustitución de aspartato (D) por glicina (G) en la posición 614] en la proteína S. Esta mutación D614G condicionó la pandemia al reemplazar a la detectada inicialmente en China, y ya en junio de 2021 era dominante a nivel mundial. Sin embargo, no se asoció a una mayor gravedad ni a pérdida de rendimiento de las pruebas diagnósticas de laboratorio, tampoco a disminución de la capacidad neutralizante de los anticuerpos ni a menor efectividad terapéutica, aunque era más infecciosa.7,15,31
El aumento en la capacidad infectante de la variante D614G no es exclusiva de seres humanos, puesto que también infecta células de otras especies, como las de murciélagos y pangolines, que expresan ACE2.15
Variantes preocupantes o de preocupación
Las VOC de SARS-CoV-2 se asocian a uno o más de los siguientes cambios en cierto grado de importancia para la salud pública mundial:10
Aumento de la transmisibilidad o cambio perjudicial en las características epidemiológicas de la COVID-19.
Aumento de la virulencia o cambio en la presentación clínica de la enfermedad.
Disminución de la eficacia de las medidas sociales y de salud pública o de los diagnósticos, las vacunas y las terapias disponibles.
Entre las VOC se encuentran la alfa, beta, gamma y delta. Para estas variantes las pruebas confirmatorias sugieren asociación a una virulencia aumentada en términos de transmisibilidad, infectividad, gravedad de la enfermedad, junto con una reducción de la eficacia de los fármacos, de los anticuerpos monoclonales; todo ello generado por una exposición previa a vacunas o por el fracaso de las técnicas diagnósticas disponibles.2)
Estos aspectos aumentan la virulencia, ejerciendo cambios adversos en las características epidemiológicas de la COVID-19.2,7,32
Por otro lado, las variantes de alta importancia (VOHC) evidencian claramente que las medidas preventivas y terapéuticas tienen una menor eficacia que las variantes previamente circulantes.2,7 Afortunadamente, por el momento no existe ninguna que se corresponda a esta categoría.
Variantes de interés
Se consideran como VOI del SARS-CoV-2 aquellas con las siguientes particularidades:10
Con cambios genéticos que se predicen o que afectan las características del virus, como la transmisibilidad, la gravedad de la enfermedad, el escape inmunológico, el diagnóstico o el escape terapéutico.
Las identificadas como causantes de una transmisión comunitaria significativa o de múltiples conglomerados de COVID-19 en varios países, con una prevalencia relativa creciente, junto con un número de casos cada vez mayor a lo largo del tiempo u otros impactos epidemiológicos aparentes que sugieran un riesgo emergente para la salud pública mundial.
Entre las VOI figuran zeta, eta, theta, iota, kappa, lambda y mu. Con los cambios evolutivos que experimenta el SARS-CoV-2 es probable que las VOI se conviertan en VOC y que aparezcan nuevos mutantes.
Variante británica o alfa
La variante británica, alfa, B.1.1.7 o 20I/501Y.V1 o VOC202012/01 se detectó en diciembre de 2020 en el Reino Unido y rápidamente se ha extendido suplantando, en muchas ocasiones, a la cepa original. El 20 de abril de 2021, la OMS comunicó su presencia en más de 130 países. Se ha convertido en la dominante debido a su mayor transmisibilidad.4,14,15,33
Esta también se asocia a una mayor gravedad y mortalidad en personas adultas. El incremento de la letalidad informado por esta cepa es de 4,1 muertes por 1000 casos detectados (en relación con la de las cepas previamente circulantes estimada en 2,5 por 1000 casos); es decir, un aumento de 35 % del riesgo de muerte.7,13,33,34 Sin embargo, no parece ser más grave en niños y jóvenes.
La variante alfa muestra una relativa resistencia a la neutralización por anticuerpos monoclonales frente al antígeno S, con bajo riesgo de reinfección; pero no es más resistente ante el plasma de individuos expuestos a la infección natural o tras la vacunación.7,33
En comparación con los virus ancestrales, alfa acumula 23 mutaciones,4,34 de las cuales 14 son no sinónimas: [T1001 IA1708D y I2230T] en el marco de lectura abierto ORF1ab, [N501Y, A570D, P681H, T716I, S982A y D1118H] en la proteína S, [Q27stop, R52I e Y73C] en el ORF8 y [D3L y S235F] en la proteína de la nucleocápside (N); seis son sinónimos: [C913T, C5986T, C14676T, C15279T y T16176C] en ORF1ab y [T26801C] en el gen M (membrana); y tres son deleciones: [SGF 3675-3677del] en ORF1ab y [H69-V70del e Y144del] en la proteína S.
La deleción H69-V70del puede disminuir la sensibilidad diagnóstica de ciertas pruebas de PCR. Sin embargo, dado que la mayoría de las técnicas de PCR combinan múltiples dianas, estos cambios no parecen repercutir sobre el rendimiento de los tests rápidos.7,15)
Respecto a los cambios en la variante alfa, 47 % ocurren en la proteína S, incluido el RBD. Estas mutaciones pueden influir en la interacción con el receptor ACE2, aumentando la tasa de infección, dañando la eficacia de los anticuerpos neutralizantes y de las células T durante la infección o vacunación, o afectando la eficacia del tratamiento.34
Variante sudafricana o beta
En diciembre de 2020, se informó en Sudáfrica la variante beta, B.1.351 o 20H/501.V2, que quizá pudo aparecer algunos meses antes y se ha extendido por diversos países. Hasta junio de 2021 representaba más de 50 % de las infecciones en muchas naciones del África.7,13,15,33
Esta variante posee 12 mutaciones no sinónimas y una deleción, al compararla con la cepa de referencia de Wuhan. Alrededor de 77 % de esas mutaciones se localizan en la proteína S [L18F, D80A, D215G, LAL 242−244 del, R246I, K417N, E484K, N501Y, D614G, y A701V], mientras que las restantes se localizan en las proteínas virales ORF1a [K1655N], la envoltura (E) [P71L] y N [T205I]. A diferencia de la cepa británica, no porta la deleción 69/70. La mutación N501Y de beta, aunque está también presente en la variante británica, parece ser filogenéticamente distinta.4,7,13,34
La mayoría de las mutaciones se producen en los dominios NTD y RBD de la proteína S, lo que sugiere que podrían escapar de los anticuerpos neutralizantes y dañar la eficacia de las vacunas en mayor cuantía que la variante alfa. Estas mutaciones pueden ayudar al virus a evadir la respuesta inmunitaria desencadenada por la infección viral previa, con riesgo de reinfección. Su rasgo más significativo es su alta tasa de transmisión.13,33,34)
De los cinco anticuerpos monoclonales aprobados en EEUU, el bamlanivimab, el etesevimab y el casirivimab son, en gran medida, inactivos contra la variante beta, mientras que el imdevimab y el sotrovimab, que se unen al núcleo del RBD, conservan la actividad neutralizante.13
Variante brasileña o gamma
La variante gamma, brasileña, P.1 o B.1.1.28.1 o 20J/501Y.V3 se detectó en Japón en viajeros procedentes de Brasil, el 6 de enero de 2021.34 El rápido incremento de ingresos hospitalarios fue un problema significativo con esta variante.
Según el informe epidemiológico de la OMS, hasta el 20 de abril de 2021 esta variante había sido detectada en 52 países. Para junio de 2021 representaba una alta proporción de infectados en algunos países de América del Sur y el Caribe y 10 % de las infecciones en EE.UU.13,33
Esta contiene 17 mutaciones no sinónimas: [L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I, and V1176F] en la proteína S, [S1188L, K1795Q, y E5665D] en ORF1ab, [E92K] en ORF8 y [P80K] en la proteína N; una deleción [SGF 3675-3677del] en ORF1ab y 4 mutaciones sinónimas. Gamma posee el mayor número de mutaciones en la proteína S.4,13,34
La mutación N501Y se presenta en las tres variantes, mientras que L18F, K417T, E484K, y D614G están en las variantes beta. Este grupo de 5 mutaciones de S tienen importantes implicaciones para la evasión de la inmunidad mediada por anticuerpos en personas vacunadas y convalecientes. Su perfil de resistencia a los anticuerpos monoclonales aprobados es similar al de la variante beta.13,34
Una forma de distinguir la variante sudafricana de la brasileña es que esta última carece de la mutación K417N, lo cual representa una amenaza contra los actuales tratamientos con anticuerpos y, en menor proporción, contra la eficiencia protectora de las vacunas.7,12,15)
Variante india o delta
La variante delta o india o B.1.627 se registró primero en el estado de Maharashtra en octubre de 2020; luego se diseminó por la India.33,34 Las secuencias virales tienen dos sustituciones críticas de aminoácidos (L452R y E484Q) en RDB de la proteína S. Esta tiene 3 sublinajes B.1.617.1, B.1.617.2 y B.1.617.3, caracterizada por la mutación L452R en S, mientras E484Q está en B.1.617.1 y B.1.617.3, pero no en B.1.617.2. La principal variante actual en Reino Unido es B.1.617.2.
Dicha cepa se ha denominado como «doble mutante» porque aúna las mutaciones L452R de la cepa de California con la mutación E484Q, aunque este término carece de sentido porque las otras variantes del SARS-CoV-2 también presentan varias mutaciones.7
El genoma presenta 17 mutaciones,34 de las cuales la D614G es compartida con otras variantes altamente transmisibles como alfa, beta y gamma, la L452R confiere mayor afinidad de la proteína S por ACE2 y una menor reacción del sistema inmunitario y la P681R puede aumentar la infectividad de la variante. De hecho, Wang et al35 encontraron que la infección con esta variante presenta mayor transmisibilidad, carga viral y riesgo de progresión de la enfermedad que la cepa “salvaje”.
Cuando la pandemia surgió en la India, dos variantes con un ancestro común: delta (B.1.617.2) y kappa (B.1.617.1), representaron una alta proporción de infecciones;13 las dos contienen la mutación L452R del RBD, la P681R del sitio de corte de la furina proximal y varias mutaciones dentro de orf3, orf7a y el gen de la nucleocápside. La variante kappa contenía la mutación E484Q, mientras delta contenía la T478K, ambas en RBD.
Las dos variantes también contienen diferentes mutaciones dentro de orf1a/b y los dominios NTD y S2 de la proteína S. Aunque E484Q tiene más probabilidades que T478K de evadir la neutralización de los anticuerpos, solo delta es más transmisible y se ha extendido a 54 países, sustituyendo a la variante alfa en el Reino Unido y los Estados Unidos.13
En el plasma de vacunados con AZD1222 las proporciones más altas mostraron una actividad neutralizante reducida contra esta variante.13 En estudios de casos y controles realizados en el Reino Unido, la vacuna BNT162b fue aproximadamente 85 % efectiva para la variante delta, mientras que la vacuna AZD1222 fue aproximadamente 60 % efectiva.
La kappa o B.1.617.1 con las mutaciones en S L452R, E484Q, D614G, P681R, también presenta un impacto en cuanto a neutralización y transmisión.34 En contraste con delta, no se ha demostrado incremento de la transmisión. Sin embargo, debido a la existencia de las mutaciones L452R y E484Q en RBD, la variante kappa tiene, de alguna manera, mayor capacidad para evadir la inmunidad humoral.
Variante californiana o épsilon
La variante épsilon (B.1.427/B.1.429) fue identificada en EE.UU. en septiembre de 2020, aunque se sugiere que el progenitor de ambos linajes surgió en mayo de 2020 y luego dio lugar a los linajes B.1.427 y B.1.429. En febrero de 2021 representaba 15 % de las infecciones en ese país., pero en junio de 2021 su prevalencia se redujo a menos de 1 % y para mayo se detectó en 34 países adicionales.11,13
La variante B.1.429 se ha extendido rápidamente, es más transmisible y menos susceptible a la neutralización por el suero de sujetos convalecientes o vacunados.34,35 Estas variantes han sido clasificadas como VOC por los CDC (Center of Disease Control and Prevention), mientras que la OMS las considera solo como VOI.7,11,33)
Los dos linajes B.1.427 y B.1.429 poseen las mismas mutaciones en S (S13I en el péptido señal, W152C en el NTD y L452R en el RBD), pero albergan diferentes mutaciones en otros genes del SARS-CoV-2.11 En conjunto, este análisis ilustra el aumento de la incidencia de B.1.427/B.1.429 y su progresiva propagación geográfica desde California hacia otras regiones de los Estados Unidos y otros países.
Según los CDC, la VOC denominada épsilon presenta las mutaciones S13I de la proteína S en el péptido señal, W152C en el NTD y L452R en RBD.11,13 El plasma de personas que recibieron una vacuna basada en el ARNm, aislado de Wuhan-1 o de convalecientes, mostró títulos neutralizantes que se redujeron entre 2 y 3,5 veces contra esta variante en relación con los pseudovirus de tipo “salvaje”. Se estimó que era 20 % más transmisible que los linajes cocirculantes y que se asociaba a niveles del virus dos veces más altos en las vías respiratorias superiores.13)
Variante lambda o C.37
La variante lambda fue notificada inicialmente en Perú en diciembre de 2020 y presenta una alta prevalencia en algunos países de América del Sur. El secuenciado reveló una deleción (D3675- 3677) en el marco de lectura (ORF1a) de tipo “salvaje”. También tiene una nueva deleción (D246-252) y múltiples mutaciones no sinónimas (G75V, T76I, L452Q, F490S, D614G, y T859N) en la proteína S. Las mutaciones L452Q y F490S están en RBD y la deleción Δ246-252 en NTD.13,34
Las mutaciones L452Q y F490S se presentan en RDB de S. La mutación F490S se asocia a disminución de la neutralización por anticuerpos. Esta variante presenta 19 mutaciones que la hacen más transmisible y más resistente a anticuerpos inducidos por la vacunación o por la exposición previa al virus.13,34)
Otras variantes
La variante eta (B.1.525) se identificó primero en Nigeria en diciembre de 2020.34 Sus mutaciones en S incluyen E484K, D614G, Q677H con impacto en la neutralización por anticuerpos y la transmisibilidad. Se encuentra en niveles bajos en muchos países, entre los cuales Nigeria presenta la mayor proporción de infecciones. Se ha estudiado poco la capacidad de los anticuerpos monoclonales, el plasma de convalecientes y el de sujetos vacunados para neutralizar esta variante.
La theta o P.3 o B.1.1.28.3, identificada en Filipinas a inicios de 2021, con impacto en la neutralización por anticuerpos y la transmisión.34 Contiene 13 mutaciones que definen el linaje, incluidas N501Y, E484K, P681H y D614G en S y la deleción de NTD en las posiciones 141-143.
La iota o B.1.526 o 20C/S: 484K, identificada primero en el estado de Nueva York, EE.UU., en diciembre de 2020, presenta las mutaciones en S: E484K, D614G, A701V e impacto en la neutralización. En junio de 2021 tenía una prevalencia en esa nación de 5-10 %, aunque es rara en otros países. Contiene la misma deleción nsp6 de las variantes alfa, beta y gamma. El plasma de aproximadamente 40 % de convalecientes y el de 30 % de los individuos vacunados con ARNm muestran una disminución de 3 a 10 veces en la actividad neutralizadora.13,34
La zeta o P.2 o B.1.1.28.2, detectada en Brasil en enero de 2021, presenta mutaciones en S: E484K, D614G e impacto en la neutralización. Las variantes iota, eta y zeta se caracterizaron primariamente por la mutación E484K en RBD y no se ha demostrado que ninguna cause una enfermedad más grave. La variante zeta era común en Brasil a finales de 2020 y principios de 2021, pero parece estar disminuyendo su prevalencia.13,34
Implicaciones clínicas de las variantes
En áreas densamente pobladas con escasas medidas de prevención o protección por falta de servicios básicos de salud o por desinterés gubernamental, el SARS-CoV-2 se difunde libremente y halla nuevos huéspedes donde replicarse.6 Así, aumenta también la probabilidad de generar mutaciones espontáneas, que pasan a ser dominantes si le aportan ventajas “competitivas” al virus.
El impacto de estas variantes en los programas globales de vacunación ha suscitado inquietud, por lo que se considera prioritario desarrollar marcadores que correlacionen vacuna y protección. Se ha planteado que las nuevas variantes podrían escapar más fácilmente a la respuesta inmunitaria.7,36)
Asimismo, en estudios en animales37,38 y seres humanos8,39,40,41 se ha obtenido una respuesta neutralizante aceptable o disminuida, inducida por la vacunación o el suero de convalecientes frente a diferentes variantes.
Hoffmann et al42 demostraron que la entrada celular de todas las variantes se bloquea por los inhibidores camostat, EK-1 y EK-1-C4. En cambio, la entrada de beta y gamma fue parcial (casirivimab) o totalmente (bamlanivimab) resistente a los anticuerpos en el tratamiento contra la COVID-19.
Las manifestaciones clínicas de estas variantes del SARS-CoV-2, y de otras que surjan en el futuro, no solo depende de las mutaciones en las proteínas vitales para la replicación viral, sino también de factores ambientales y relacionados con estilos de vida, como la nutrición, las enfermedades asociadas, los hábitos tóxicos, la contaminación ambiental y la higiene personal y comunitaria, además de factores genéticos que pudieran contribuir a la transmisibilidad, virulencia y evasión de la respuesta inmunitaria humoral y celular del SARS-CoV-2. Es difícil desligar estos factores que interactúan estrechamente en una compleja red.7,10,14,43 En el cuadro se resumen las variantes genéticas del SARS-CoV-2.
Conclusiones
Las mutaciones son sucesos comunes en el proceso de replicación de los virus, lo que evidentemente también ocurre en el SARS-CoV-2, que posee nuevas variantes de características epidemiológicas diferentes, que han alarmado a la comunidad científica internacional por la posibilidad de una mayor transmisibilidad y letalidad, además de su evasión a los mecanismos inmunitarios naturales y adquiridos, lo cual puede incrementar el riesgo de reinfección, provocar mayor gravedad de la enfermedad y reducir la eficacia de los medicamentos antivirales y de las vacunas.
Entre las estrategias propuestas para combatir las variantes del virus se encuentran el aumento de las dosis y la alternancia de vacunas, así como el desarrollo de vacunas específicas frente a diferentes variantes.

