










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La displasia fibrosa (DF) fue descrita por Lichtenstein y Jaffe como un desorden esquelético. El tejido fibroso en expansión, se produce en la porción esponjosa de los huesos afectados. La matriz fibrosa del esqueleto no presenta osteoblastos; sin embargo, aparecen escasas y defectuosas trabéculas generadas por metaplasia del tejido fibroso.1) Las trabéculas son de estructura plexiforme y no laminillar.
La DF puede afectar un hueso (monostótica) o varios huesos (poliostótica). Su manifestación más común es en la infancia o al inicio de la adolescencia. La DF poliostótica puede relacionarse con la pubertad temprana y áreas de pigmentación cutánea.
Su etiología se debe a un desequilibrio en la función de las células osteogénicas, responsable de importantes deformaciones del esqueleto.2 La DF es una enfermedad genética no hereditaria atribuida a una mutación sin sentido del gen GNAS1 (del inglés, guanine nucleotide-binding protein, alpha-stimulating activity peptide 1).3,4) Esta mutación provoca una proliferación anormal y diferenciación de los pre-osteoblastos; aunque los cambios malignos son relativamente bajos.
Radiográficamente, las lesiones de DF se caracterizan por un aspecto de vidrio deslustrado; debido a la mezcla de elementos óseos y fibrosos. Puede ser unilocular o multilocular y, generalmente, está asociada con la expansión de la cortical ósea. El mejor método para identificar la DF es la tomografía computarizada; ya que permite determinar la localización, extensión de la lesión y es esencial en la planificación del tratamiento y en el procedimiento quirúrgico.5
Las manifestaciones clínicas se derivan del desplazamiento de las estructuras vecinas debido al crecimiento progresivo tumoral; el cual varía la localización y afecta el maxilar, cuando su aumento de volumen es indoloro, unilateral, lento y progresivo. Produce asimetría facial, cuando compromete el hueso temporal con pérdida de la audición. Si el frontal, el esfenoides y etmoides son los huesos comprometidos, el resultado será obstrucción nasal.6,7,8)
La forma monostótica comprende el 70% de los casos y es igual de frecuente en ambos sexos. Generalmente, detiene su crecimiento durante la adolescencia, cuando cierran las epífisis.8,9,10 Por lo poco frecuente de su aparición, se decide la realización de este reporte.
Presentación de Caso
Paciente masculino de la raza blanca de 56 años de edad que acude al Servicio de Imagenología del Hospital Lenin, de Holguín, Cuba, remitido de la consulta de Oftalmología, que refiere cefalea y trastornos visuales de largo tiempo de evolución.
En el examen físico del paciente se constata asimetría facial a nivel del hueso frontal con proptosis del globo ocular derecho (Figura 1).

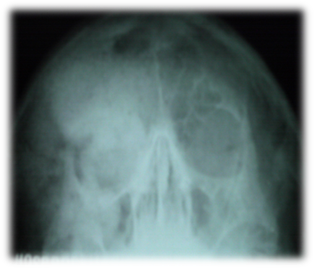
Como parte del estudio imagenológico, se realiza una radiografía de senos paranasales (vista de Water) (Figura 2). En dicha radiografía se observa una opacidad con aspecto de vidrio esmerilado que ocupa el hueso frontal y las celdillas del seno frontal derecho, con ausencia de neumatización que, compromete la órbita derecha, el hueso maxilar superior y seno maxilar derecho.
Para complementar el estudio y precisar la extensión de la afección, se le indica al paciente una tomografía axial computarizada de senos paranasales (Figura 3).

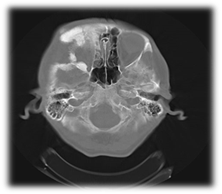
En la tomografía axial computarizada de senos paranasales, con espesor de cortes de 2 mm, se observa un marcado engrosamiento y esclerosis con aspecto de vidrio esmerilado que expande la cortical y deforma la arquitectura ósea normal; que interesa al hueso frontal derecho, maxilar superior, arco cigomático, hueso esfenoide y piso de la fosa media, y reduce el diámetro orbitario; que ocasiona exoftalmos ipsilateral y seno frontal, con ausencia de neumatización en celdillas derechas. Se comparan con las estructuras contralaterales, las cuales no muestran alteraciones. Se concluye como una displasia fibrosa monostótica (Figura 4).
Discusión
La DF es una enfermedad ósea benigna, causada por mutaciones somáticas del gen GNAS1, que codifica la subunidad Gsα, cuyas manifestaciones clínicas aparecen en edades tempranas de la vida. Las lesiones dejan de crecer en la adultez; la enfermedad exhibe un mosaisismo de mutaciones somáticas.4
El diagnóstico presuntivo de DF de primera instancia es difícil; porque aborda todas las posibles enfermedades que podrían presentarse de forma similar. En primer lugar, es necesario diferenciar dentro del grupo de enfermedades infrecuentes aquellas con mayor incidencia y tomarlas en consideración; así como revisar las de curso benigno y maligno, sin dejar fuera aquellas lesiones del hueso primarias o secundarias. El diagnóstico definitivo depende de la toma de la biopsia junto a los estudios anatomopatológico, endocrinológico, y la gammagrafía ósea, para identificar otras lesiones insospechadas.4,6,8)
Las opciones terapéuticas deben ser evaluadas en cada caso, después de valorar los factores relacionados con el paciente, su enfermedad, la disponibilidad de medicamentos y la tecnología de avanzada. Hasta la fecha, se continúa trabajando en la aplicación de un tratamiento médico específico para su curación. Se recomienda el procedimiento quirúrgico, en aquellos casos, en los que sea posible realizar la excéresis completa de la lesión para evitar recidivas. Esta también se indicada en pacientes con manifestaciones clínicas deficitarias, asociadas al efecto compresivo de las lesiones o cuando la lesión afecta la estética facial y la función, como en este caso.9,10,11

