










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El cáncer es un grupo de enfermedades en que las células no responden a la restricción normal al crecimiento y presentan capacidad de invadir o diseminarse a otras partes del organismo. (1
La magnitud del cáncer como problema de salud es enorme, tanto por su costo humano y el impacto económico que representa para el sistema de salud; en Cuba, el cáncer fue la primera causa de muerte en el sexo masculino durante el año 2017, y la segunda en el sexo femenino.2En el caso de las provincias, Pinar del Río, Artemisa, Ciego de Ávila, Camagüey,
Las Tunas y el municipio especial de Isla de la Juventud presentaron el cáncer como primera causa de muerte en ambos sexos, relegando a las enfermedades cardiovasculares al segundo lugar.
La provincia de Holguín presentó similar mortalidad total de cáncer y enfermedades cardiovasculares. (2
La mortalidad por cáncer ha ido aumentando sostenidamente durante los últimos 25 años. 3) El riesgo real de morir por cáncer en Cuba tuvo un incremento del 78,21% entre los años 1970 y 2006. En el año 2006 la tasa cruda de mortalidad por cáncer casi duplicó la existente al inicio de la década del 70. 3) Las tasas de mortalidad muestran un ligero decrecimiento, mientras que las de incidencia, muestran una tendencia francamente ascendente.
En general en Latinoamérica y en particular en el Ecuador, las tasas de prevalencia y mortalidad siguen un comportamiento similar al de Cuba.
Al cierre de 2014 la tasa mortalidad más elevada por tipos de cáncer en Ecuador, en el sexo masculino correspondió a próstata, estómago, pulmones, hígado y colorrectal, mientras en mujeres predominó el cervicouterino, estómago, mama, hígado y colorrectal. 4 En ambos sexos las muertes por otros tipos de cánceres fueron en hombres y mujeres de 39,1% y 46,6%, respectivamente.
Una de las características del cáncer es que su incidencia y mortalidad aumenta marcadamente con la edad. Estas comienzan a incrementarse a partir de los 10 años de edad, lo que puede tener relación con el tiempo requerido para que las células acumulen las mutaciones que provocan las múltiples etapas en el desarrollo del cáncer.
Con el proceso de envejecimiento poblacional las probabilidades de enfermar y morir por cáncer se elevarán, a lo que contribuirá el cambio climático; esto incrementará la demanda de recursos humanos y materiales, con el aumento de los costos asociados al diagnóstico, tratamiento, seguimiento y rehabilitación de los pacientes, con el consecuente aumento en la supervivencia y la prevalencia de casos, asociado al uso de nuevas posibilidades terapéuticas con cobertura nacional.
Se ha encontrado que la incidencia de cáncer es determinada en gran parte por el medio y hábitos de vida, por lo que, en principio, la mayoría de los cánceres puede ser prevenible. En Ecuador se realiza monitorización del cáncer con registro oncológico hospitalario y se aplican políticas de prevención primaria del cáncer que actúan sobre los principales factores de riesgo de la enfermedad como el hábito de fumar, el sedentarismo y el sobrepeso.4
Los objetivos de esta revisión fueron describir los mecanismos moleculares que provocan la transformación cancerosa y los enfoques terapéuticos basados en los mecanismos moleculares que causan la enfermedad. El conocimiento de estos aspectos tiene repercusión sobre el diagnóstico, pronóstico y tratamiento de las neoplasias malignas.
Desarrollo
Métodos de búsqueda de información
Se realizó la revisión bibliográfica en PubMed con los descriptores mecanismos moleculares del cáncer, oncogenes, proto-oncogenes y genes supresores de cáncer. La búsqueda abarcó desde el 2000 hasta el 2019, en la que se encontraron 3 933 662 artículos en PubMed. En una segunda etapa se seleccionaron contribuciones que trataran los aspectos señalados en los descriptores, así como diagnóstico, prevención y terapéutica.
Se eliminaron las contribuciones duplicadas en las búsquedas y se seleccionaron las publicaciones más representativas, que se organizaron en una carpeta en una computadora personal. Además, se revisaron algunas revistas médicas cubanas y libros de diversas especialidades de autores reconocidos, con el objetivo de obtener una visión más integradora del tema.
Genética del cáncer
El cáncer es una enfermedad genética. El hecho de que la progresión del cáncer está dirigida por una secuencia de mutaciones somáticas en genes específicos ha ganado aceptación en los últimos 25 años.
Estas alteraciones genéticas son específicas para cada tumor, pero existen características comunes para los genes que causan cáncer. Debemos tener presente que hay diferencias entre las mutaciones somáticas (presentes en el cáncer, pero no en el resto del organismo) y las mutaciones de la línea germinal (heredadas y presentes en todas las células del organismo). 6
De esta forma, las lesiones genéticas no letales constituyen la base de la carcinogénesis y pueden producirse por la acción de agentes ambientales, que incluye tanto los agentes exógenos (sustancias químicas, radiación o virus), como los productos endógenos del metabolismo celular; pero también pueden ser heredadas en la línea germinal. Hay que señalar que no todas las mutaciones son inducidas por el medio ambiente, algunas son espontáneas y estocásticas.7
El estudio de los tipos de mutación, así como las consecuencias de las mismas en las células tumorales se conoce actualmente como genética del cáncer.8
La gran mayoría de los cánceres se origina de una sola célula, es decir, tiene origen clonal. Se necesitan varias mutaciones acumulativas para que una célula normal adquiera un fenotipo maligno. El proceso puede considerarse una micro evolución darwiniana en la cual, cada mutación produce una célula con ventaja de crecimiento, lo que provoca mayor representación con respecto a sus células vecinas. Basado en la frecuencia con que el cáncer se incrementa con la edad y estudios de genética molecular, se considera que son necesarias entre 5 y 10 mutaciones acumuladas para originar una célula maligna.1,8,9
Se han determinado cambios fundamentales en la fisiología de estas células que les confieren su fenotipo maligno, todos los cuales representan, a través de diversos mecanismos, consecuencias de los cambios moleculares producidos por las mutaciones. Estos cambios son: 1,5,6
Autosuficiencia en señales de crecimiento: proliferan sin estímulos externos.
Insensibilidad a las señales inhibidoras del crecimiento, como el factor de transformación del crecimiento beta (TGF-β) y los inhibidores de las quinasas dependientes de ciclinas (CDKIs). Las células normales responden a señales como el contacto con otras células (inhibición por contacto), que detienen la proliferación.9,10
Evasión de la apoptosis: son resistentes a la muerte celular programada, como consecuencia de la inactivación de p53 o la activación de genes antiapoptósicos.10,11
Potencial replicativo ilimitado: no tienen restricciones a su capacidad proliferativa, evitando la senescencia celular y la catástrofe mitótica, es decir, evaden el límite de Hayflick.
Angiogénesis mantenida: las células cancerosas estimulan la angiogénesis mediante la secreción de VEGF (factor de crecimiento vascular endotelial), FGF (factor de crecimiento de los fibroblastos) e IL-8 (interleucina 8), ya que para poder proliferar necesitan el suministro de nutrientes y oxígeno, así como la remoción de los productos de desecho de su metabolismo. Además, no necesitan soporte estructural, como la matriz extracelular (pérdida de la dependencia de anclaje). 10
Habilidad de invadir y metastizar: las metástasis constituyen la causa de la mayoría de las muertes ocasionadas por cáncer (proteína CD36).12-14
Defectos en la reparación del DNA: existen defectos en la reparación del DNA debido al daño causado por los carcinógenos o producidos durante la proliferación celular incontrolada, lo que ocasiona inestabilidad genómica 15,16) y mutaciones en los proto-oncogenes y los genes supresores tumorales.
Oncogenes
Existen dos tipos de genes de cáncer: oncogenes y genes supresores de cáncer. Ambos tipos ejercen sus efectos a través del control de la división o la muerte celular mediante mecanismos complejos.
Los proto-oncogenes controlan el crecimiento y la división celular normales. Estos genes codifican factores de crecimiento, receptores de factores de crecimiento, proteínas de transducción de señales, factores de transcripción, reguladores del ciclo celular y reguladores de la apoptosis.
Los proto-oncogenes se convierten en oncogenes por mutaciones en el DNA que causan ganancia de función, lo que produce, bien una proteína que funciona en ausencia de los eventos activadores normales o una cantidad aumentada de la proteína normal. Estas mutaciones generalmente, ocurren en un solo alelo del oncogén y presentan un comportamiento dominante. Los oncogenes ejercen un efecto positivo sobre la formación tumoral (tabla I).
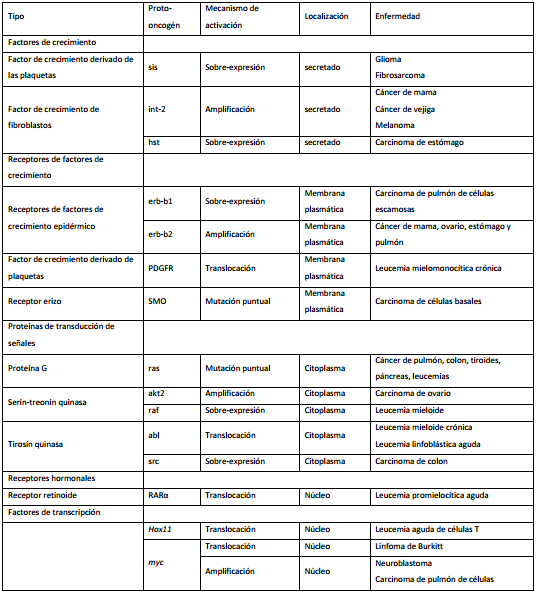
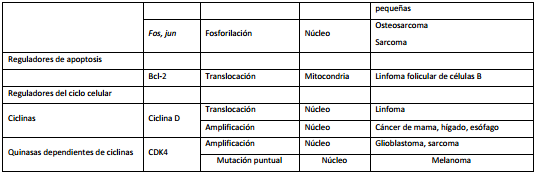
Se conocen varios mecanismos por los cuales los proto-oncogenes se convierten en oncogenes y se activan.
Mutación puntual
Entre las causas de estas mutaciones se encuentran carcinógenos químicos del medio ambiente (benceno) o que pueden ser ingeridos y activados por el metabolismo del organismo (dimetilnitrosamina), agentes quimioterapéuticos, que, aunque están diseñados para eliminar las células proliferativas, también pueden provocar nuevas mutaciones y tumores mientras se eliminan los anteriores, la luz ultravioleta y las radiaciones ionizantes, entre otros. 1
Las proteínas ras, son proteínas G que participan en la transducción de señales; las mutaciones en uno de los genes RAS, (HRAS, KRAS O NRAS) están presentes en un 85% del cáncer de páncreas, 45% del cáncer de colon y entre 30 y 50% del cáncer de pulmón, pero son menos comunes en otros tipos de cáncer, aunque pueden tener frecuencia significativa en la leucemia, y cáncer de tiroides.1,21,23
Es de señalar, en contraste con la diversidad de mutaciones encontradas en los genes supresores, que la mayoría de los genes RAS activados, contienen mutaciones puntuales en los codones 12, 13 o 61, lo que disminuye la actividad GTPasa, provocando la activación constitutiva de la proteína RAS mutante, que a su vez activa la proteín quinasa A, lo que ilustra el importante papel que desempeñan las proteín quinasas en la señalización de los procesos relacionados con la división celular (fig. 1). 24
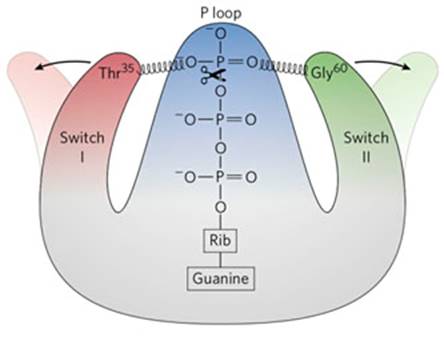
Fig.1. Se muestran los cambios conformacionales en la proteína ras. Al perder la acción GTPasa, (marcada por la tijera), continúan las interacciones entre la treonina 35, la glicina 60 y el grupo fosfato del GTP, lo que impide el cambio de conformación de los interruptores I y II para inactivarla. (tomado de Lehninger Principles of Biochemistry, 7th edition)
De esta forma, se han desarrollado varios inhibidores de quinasas específicas, como el mesilato de imatinib, que logra remisión en estadios precoces de la leucemia mieloide crónica mediante la inhibición de la tirosín quinasa de Abl. 24
El patrón de mutaciones restringido observado en los oncogenes, comparado con el de los genes supresores, refleja que las mutaciones con ganancia de función son menos probables que aquellas que produce la pérdida de actividad. 25
Esto se debe, a que la inactivación de un gen puede ocurrir mediante la introducción de un codón de terminación, mientras que la activación necesita sustituciones precisas en residuos que provocan el aumento de la actividad de la proteína codificada.
Es importante señalar que la especificidad de las mutaciones de oncogenes proporciona oportunidades diagnósticas, ya que las pruebas que identifican mutaciones en posiciones definidas son más fáciles de diseñar que las pruebas que detectan cambios aleatorios en un gen.
Amplificación del DNA
El segundo mecanismo para la activación de los oncogenes es la amplificación de una secuencia de DNA, que provoca sobreexpresión del producto génico. Este aumento en las copias del DNA, puede causar alteraciones cromosómicas citológicamente identificables, como son las regiones de teñido homogéneo (HSRs), si están integradas en los cromosomas o dobles mínimas, si son extra cromosómicas. Actualmente existen técnicas que permiten explorar el genoma completo en búsqueda de ganancias o pérdidas de secuencias de DNA.
Se han encontrado numerosos genes amplificados en el cáncer. Algunos de ellos, incluidos CMYC y LMYC, fueron identificados por su presencia dentro de secuencias amplificadas de DNA de tumores por su homología con oncogenes conocidos. Se asocia a cáncer de pulmón de células pequeñas y se estudia la terapéutica epigenética combinando inhibidores de la metiltransferasa del ADN con inhibidores de la desacetilación de las histonas. 18
Debido a que la amplificación en algunos cánceres, particularmente en los sarcomas, incluye cientos de miles de bases, pueden encontrarse muchos oncogenes. En algunos tipos de sarcomas se han encontrado simultáneamente amplificados MDM2, GLI, CDK4 y SAS en la posición 12q13-15.
La amplificación de genes celulares es frecuentemente un predictor de mal pronóstico, por ejemplo, ERBB2/HER2 y NMYC se encuentran frecuentemente amplificados en el cáncer de mama y el neuroblastoma, respectivamente.
El gen c-Myc codifica un factor de transcripción, que es una proteína multifuncional que participa en la regulación del ciclo celular, la apoptosis y la transformación celulares. También se han encontrado alteraciones de Myc en los carcinomas de cérvix, colon, mama, pulmones y estómago. El incremento de la activación por translocación, aunque no es universal, juega un importante papel en la progresión maligna.
Los cambios epigenéticos, mediante la utilización de agentes demetilantes del ADN combinado con inhibidores de la desacetilación se han utilizado para mejorar la respuesta inmune al cáncer mediante la depleción mantenida de MYC. 18
La sobreexpresión del receptor del factor de crecimiento epidérmico hace que en ocasiones este receptor se dimerice sin la unión de dicho factor, lo que induce al crecimiento y división celulares. Se han diseñado anticuerpos monoclonales contra los dominios extracelulares de estos receptores. El cetuximab se une al receptor en el cáncer colorrectal, lo que impide la unión del factor de crecimiento epidérmico y bloquea, además, la dimerización del receptor. Todo esto elimina el estímulo al crecimiento y división celulares.
En el 30% del cáncer de mama se sobreexpresa un receptor para el factor de crecimiento epidérmico de la familia HER2, para el que se ha diseñado otro anticuerpo monoclonal, trastuzumab o herceptina. Actualmente se realiza pesquisaje de la sobreexpresión de HER2 para efectuar el tratamiento con herceptina.25
Reorganización de los cromosomas
Las alteraciones cromosómicas en los tumores sólidos humanos como carcinomas son heterogéneas y complejas y ocurren como resultado de la frecuente inestabilidad de los cromosomas observada en estos tumores. (27-29) En contraste, las alteraciones cromosómicas en los tumores mieloides y linfoides son frecuentemente simples translocaciones, por ejemplo, transferencias recíprocas entre los brazos de un cromosoma y otro. Consecuentemente, muchos análisis detallados han sido realizados en los cánceres hematopoyéticos.
La tabla II muestra algunos ejemplos de reordenamientos de los cromosomas y los genes asociados o desregulados por el mismo.
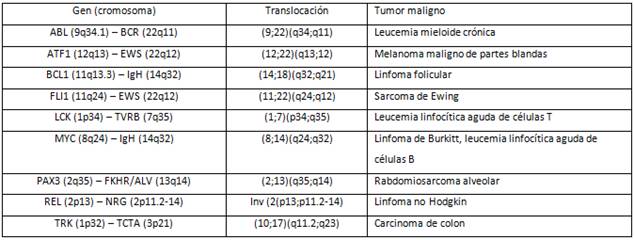
Las translocaciones son comunes en los tumores linfoides debido a que este tipo celular tiene la capacidad de reorganizar su DNA para generar receptores de antígenos (recombinación somática). De esta forma los genes receptores de antígenos se encuentran frecuentemente involucrados en las translocaciones, lo que implica que un defecto en la regulación del receptor reorganizado puede estar involucrado en la patogénesis. Un ejemplo interesante es el linfoma de Burkitt, un tumor de células B, caracterizado por una translocación recíproca entre los cromosomas 8 y 14. El análisis molecular de los linfomas de Burkitt demuestra que los puntos de ruptura se encuentran dentro o cercanos al locus MYC en el cromosoma 8 y dentro del locus de la cadena pesada en el cromosoma 14, lo que provoca la activación transcripcional de MYC, que ahora se encuentra amplificado. 26
Se ha descubierto que la proteína YAP (del inglés, yes-associated protein), constituyente de un sistema regulatorio tanto nuclear como citoplasmático, genera una respuesta de acuerdo a la rigidez de la matriz extracellular, tensiones y áreas de adhesión mediante mecanismos de fosforilación, lo cual activa factores de transcripción.29) También se han identificado otros mecanismos regulatorios de tipo mecánico.30,31
Además de los factores de transcripción y las moléculas de transducción de señales, la translocación puede resultar en la sobreexpresión de proteínas reguladoras del ciclo celular como las ciclinas y las proteínas que regulan la muerte celular.
La primera anormalidad cromosómica detectada en un cáncer humano fue el cromosoma Filadelfia, generado por la translocación recíproca que involucra el oncogén ABL del cromosoma 9, que codifica una tirosín quinasa y se coloca en la proximidad del gen BCR, que es la región de ruptura en el cromosoma 22. La consecuencia de la expresión del producto BCR-ABL es la activación de la vía de transducción que provoca crecimiento celular independientemente de las señales externas. El imatinib, droga que bloquea la actividad de tirosín quinasa de Abl ha mostrado eficacia en el tratamiento de la leucosis mieloide crónica con muy poca toxicidad.
Inestabilidad cromosómica en los tumores sólidos
Los tumores sólidos son generalmente aneuploides y contienen un número anormal de cromosomas que muestran alteraciones estructurales: translocaciones, deleciones y amplificaciones. Estas anormalidades son conocidas como inestabilidad cromosómica. 27-29Las células normales tienen varios puntos de control que verifican los requerimientos de calidad de los procesos concluidos antes de que sea permitida la realización de los eventos posteriores al mismo.
El punto de control mitótico, que garantiza la unión apropiada del cromosoma al uso mitótico antes de permitir que las cromátides se separen, está alterado en algunos cánceres y se han encontrado tanto inhibición como sobreactivación del mismo.
Actualmente es posible la medición del número de alteraciones cromosómicas presentes en los tumores mediante técnicas citogenéticas y moleculares, lo que resulta útil para el pronóstico. Debido a que el punto de control mitótico es esencial para la viabilidad celular, puede constituir un blanco para nuevos enfoques terapéuticos.7
Genes supresores tumorales
Los genes supresores tienen efecto negativo sobre la formación tumoral, generalmente reprimen el crecimiento celular y su función se pierde en el cáncer. Debido a la estructura diploide de las células de los mamíferos, ambos alelos deben estar inactivados para que se pierda completamente la función de un gen supresor tumoral. El estudio del retinoblastoma 10,33) ha apoyado la hipótesis de dos lesiones, en que ambas copias de un gen supresor tumoral deben estar inactivadas para que ocurra el cáncer.
Hay un tipo de genes supresores tumorales, los vigilantes del genoma, que, en lugar de controlar el crecimiento celular directamente, controlan la capacidad de la célula para mantener la integridad de su genoma, como el que codifica la proteína p53. 34) Las células deficientes en los mismos, presentan una frecuencia de mutaciones mayor, que incluye oncogenes y genes supresores tumorales.
Este fenotipo mutador fue propuesto por Loeb para explicar cómo las múltiples mutaciones requeridas para el desarrollo del tumor pueden ocurrir en la vida de un individuo. Este fenotipo se ha observado en algunas formas de cáncer, algunas asociadas con deficiencias en la reparación de las bases desapareadas, como en el xeroderma pigmentosum. 35) Sin embargo, la gran mayoría de los cánceres no presentan deficiencias en la reparación y sus frecuencias de mutación son similares a las de células normales. Parecen presentar una forma diferente de inestabilidad genética 16,17) afectando la pérdida o ganancia de partes de cromosomas.
Inactivación de los genes supresores en el cáncer
La primera indicación de la existencia de genes supresores se obtuvo de experimentos en que la fusión de células cancerosas de ratón con fibroblastos normales de ratón producía un fenotipo no maligno en las células fusionadas. El papel normal de los genes supresores tumorales es restringir el crecimiento celular y la función de estos genes se encuentra inactivada en el cáncer.
Las dos lesiones somáticas observadas en los genes supresores tumorales durante el desarrollo del tumor son las mutaciones puntuales y las grandes deleciones.
El silenciamiento de genes, cambio epigenético que lleva a la pérdida de la expresión genética, ocurre conjuntamente con la hipermetilación del promotor y la desacetilación de las histonas, lo que constituye otro mecanismo de inactivación de los genes supresores tumorales.36
La modificación epigenética, es un cambio en el genoma, heredable por esa progenie celular, que no involucra una modificación en la secuencia del DNA. La inactivación del segundo cromosoma X en las células femeninas es un ejemplo de silenciamiento epigenético que previene la expresión genética del cromosoma inactivado.36
Durante el desarrollo embrionario regiones de los cromosomas de cada progenitor son silenciadas y la expresión genética se realiza utilizando el cromosoma del otro progenitor. Para muchos genes, la expresión ocurre en los dos alelos o aleatoriamente en uno de los alelos.
El papel de los mecanismos de control epigenéticos en el cáncer humano no está claro, sin embargo, la disminución general en el nivel de metilación del DNA ha sido un cambio común en el cáncer. En adición, numerosos genes, incluyendo algunos genes supresores tumorales, se encuentra hipermetilados durante la formación del tumor.
VHL y p16INK4 son ejemplos bien estudiados de genes supresores tumorales. Los mecanismos epigenéticos pueden ser responsables de reprogramar la expresión de un gran número de genes en el cáncer y de conjunto con la mutación de genes específicos, parecen ser cruciales en el desarrollo de los tumores malignos en el ser humano.
El uso de fármacos que puedan revertir los cambios epigenéticos en las células cancerosas pudiera representar una opción terapéutica en ciertos cánceres o condiciones premalignas, por ejemplo, los agentes desmetilantes (azacitidina o decitabina) han sido aprobados para el tratamiento de pacientes con alto riesgo de síndrome mielodisplástico.7
Síndromes familiares de cáncer
Una pequeña cantidad de cánceres ocurre en pacientes con predisposición genética. En estas familias, los individuos afectados presentan una mutación de pérdida de función en uno de los alelos de un gen supresor tumoral.
Los tumores en estos pacientes se deben a la pérdida del alelo normal como resultado de eventos somáticos (mutaciones puntuales o deleciones), de acuerdo con la hipótesis de las dos lesiones. De esta forma, la mayoría de las células de un individuo con una mutación de pérdida de función en un gen supresor tumoral son funcionalmente normales, y solo las pocas células que desarrollan una mutación en el alelo normal restante tendrán regulación incontrolada.8
Está establecido que los genes mutados en los síndromes familiares pueden también ser blanco de mutaciones somáticas en tumores esporádicos (no heredados). El estudio de los síndromes relacionados con el cáncer ha proporcionado información de los mecanismos de progresión de muchos tipos de tumores.
En particular, el estudio del cáncer de colon hereditario ilustra claramente la diferencia entre dos tipos de genes supresores tumorales: porteros (gatekeepers), que controlan la proliferación celular, lo que afecta el crecimiento de los tumores y los conserjes (caretakers), que son responsables de la integridad del genoma y cuando están mutados producen inestabilidad genética 16,17) e intervienen indirectamente en el crecimiento tumoral.17,37,38
La inestabilidad genética puede generarse por mutaciones individuales en un gen concreto, por pérdida o ganancia de cromosomas o, lo que parece ser más frecuente, por reorganizaciones a gran escala de los cromosomas.17
La poliposis adenomatosa familiar es un síndrome canceroso con carácter dominante ocasionado por mutaciones en la línea germinal. Se encuentra afectado el gen supresor tumoral de la poliposis adenomatosa familiar (APC) en el cromosoma 5. Los pacientes desarrollan cientos o miles de adenomas en el colon, algunos adquirirán anormalidades y una fracción de los mismos desarrollará cáncer.
Cada uno de ellos ha perdido el alelo normal remanente del APC, pero todavía no ha acumulado las mutaciones adicionales para generar células malignas.8
El gen APC es considerado un guardián para la carcinogénesis del colon: en ausencia de mutación de este gen o de otro que actúe dentro de la misma vía, el cáncer colorrectal no se puede formar. Los defectos en este proceso pueden provocar acumulación de células que normalmente deben sufrir apoptosis.8) La fig. 2 ilustra las mutaciones somáticas progresivas en el carcinoma de colon.
Algunos síndromes hereditarios asociados al cáncer de mama y otros tipos de cánceres se reflejan en el trabajo de Miguel-Soca et al.39,40Con frecuencia los factores relacionados con los estilos de vida como el hábito de fumar, las radiaciones y los hábitos dietéticos, son más importantes en la predisposición al cáncer, aunque en los tumores hereditarios los genes tienen un papel fundamental.39
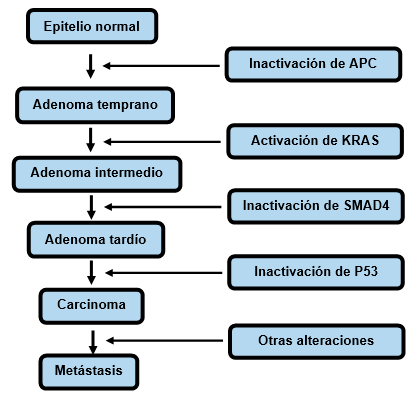
La acumulación de alteraciones en diferentes genes conduce a la progresión de un epitelio normal al carcinoma. La inestabilidad genética acelera la progresión al incrementar la probabilidad de mutación en cada etapa. Modificado de Harrison´s Principles of Internal Medicine. Chapter 101a, 2015.
Conclusiones
El cáncer es el resultado de lesiones genéticas no letales producidas por la acción de agentes exógenos (sustancias químicas, radiación o virus), así como de algunos productos endógenos del metabolismo celular. Este tipo de lesión genética también puede ser heredada.
La gran mayoría de los cánceres se origina de una sola célula, es decir, tiene origen clonal. Actualmente se conoce que son necesarias varias mutaciones acumulativas para que una célula normal adquiera un fenotipo maligno.
A medida que se esclarecen los mecanismos moleculares involucrados en esta transformación, se crean las bases para el desarrollo de nuevos enfoques terapéuticos.
La investigación sobre los mecanismos de control epigenético en el cáncer constituye un área que puede aportar procedimientos terapéuticos en algunos tipos de cáncer.

