










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La artritis reumatoide (AR) es una enfermedad sistémica inflamatoria, autoinmune, multifactorial, que involucra al sistema inmune y culmina en la destrucción articular y discapacidad, por su curso progresivo y crónico. 1 También afecta otros órganos y sistemas y su principal causa de mortalidad es la enfermedad cardiovascular. 2
Las citoquinas o citocinas son proteínas o glicoproteínas de bajo peso molecular con una vida media corta, liberadas principalmente por las células del sistema inmune, que intervienen en la transmisión intracelular de señales y la inflamación.3 Sus efectos paracrinos y autocrinos regulan la activación, la migración, la supervivencia y la diferenciación celulares. 3
Los sellos distintivos de la sinovitis en la AR son la proliferación de fibroblastos sinoviales, la formación de nuevos vasos sanguíneos y el reclutamiento de leucocitos, que conlleva a la hipertrofia de la membrana sinovial y daño del cartílago y hueso.4 Las citoquinas también intervienen en el mantenimiento de estas lesiones.
Entre las citoquinas, el factor de necrosis tumoral alfa (TNF-α) parece tener una posición dominante durante la fase inflamatoria de la enfermedad, al promover la activación de los osteoclastos, condrocitos, endotelio vascular y fibroblastos y de manera directa intervienen en la hipertrofia sinovial, además con un incremento adicional del daño por la síntesis excesiva de otras citoquinas proinflamatorias.
Otras citoquinas implicadas en el daño articular son las interleuquinas (IL): IL-1, IL-6 y GM-CSF, las dos últimas con funciones en la activación de las células T y en la diferenciación de macrófagos y células dentríticas. 4 Además, IL-6 y TNF-α ejercen potentes efectos sistémicos que favorecen algunas comorbilidades en la AR.
Otra propuesta sobre la patogenia de la AR sugiere que con la activación de los leucocitos sinoviales se liberarían citoquinas proinflamatorias y radicales libres (estrés oxidativo), que inician la inflamación y atraen a otras células inmunológicas al sitio. 1 Luego, estas especies reactivas intervienen en la destrucción del cartílago y contribuyen a la sinovitis proliferativa.
Participa en la AR una red bidireccional entre el sistema neuroendocrino y el sistema inmune, vinculada por las adipoquinas. 5 Las adipoquinas, citoquinas liberadas por los adipocitos, actúan como moléculas señales en la interacción neuroendocrina-inmune y también se sintetizan por otras células dentro de las articulaciones.
Algunos plantean en la fisiopatología de la AR la presencia de trastornos en la inmunidad adaptativa, con una hiperactividad de las células T y disfunción de las células B. 6 Estos trastornos producen defectos en la señal de transducción y la sobreexpresión de genes de citoquinas proinflamatorias, como el TNF-α, IL-1ß, IL-6, IFN-γ, por poner algunos ejemplos. Cuando los linfocitos T se activan comienzan a secretar muchas de estas citoquinas.
La AR como artritis inflamatoria es una enfermedad patogénicamente compleja, en la que intervienen vías dependientes e independientes de anticuerpos. 7 La artritis de larga data puede tener adicionales mecanismos de perpetuación de la enfermedad, aun si la enfermedad se originó por autoanticuerpos (autoinmune).
Estos conocimientos se aplican en el diseño de nuevos fármacos que se utilizan en la práctica médica cada vez con mayor frecuencia y están avalados por ensayos clínicos aleatorios y estudios epidemiológicos.
Esta revisión pretende describir el papel de las citoquinas en la fisiopatología de la AR, sin desconocer que todavía quedan aspectos no bien aclarados y polémicos.
Desarrollo
Métodos
En US National Library of Medicine National Institutes of Health (https://www.ncbi.nlm.nih.gov/pubmed), con el descriptor rheumatic arthritis, se encontraron 842 artículos científicos en todas las bases de datos de los últimos 5 años.
En ClinicalKey, de la editorial Elsevier (https://www.clinicalkey.es) con los descriptores artritis reumatoide y citoquinas, se encontraron 18 344 trabajos: en español 688, el resto en inglés.
SciELO - Scientific Electronic Library Online (https://scielo.org/es), con el descriptor artritis reumatoide, se encontraron 410 referencias. Cuando se filtraron, a partir de 2014, se hallaron 146 artículos.
Artritis reumatoide
La AR es una enfermedad crónica inflamatoria autoinmune caracterizada por sinovitis que provoca destrucción del cartílago y hueso con deformidades de las articulaciones. 8 Otras características son hiperplasia, formación de autoanticuerpos y trastornos sistémicos, entre los cuales la inflamación crónica y la erosión ósea son las más frecuentes. 8
La AR es una enfermedad de etiología multifactorial, donde interaccionan factores ambientales con factores genéticos hereditarios, que establecen vínculos complejos sujetos a intensa investigación en la actualidad.
En la AR, la sinovial inflamada contiene células T, fibroblastos sinoviales y macrófagos que producen citoquinas proinflamatorias, como TNF-α, IL-1, IL-6, IL-17 y el factor estimulante de colonias de macrófagos y granulocitos (GM-CSF). Estas citoquinas activan los osteoclastos y la destrucción ósea.9
En la AR se produce una excesiva respuesta inmune a las células T. Las células T CD4+ comprenden las células T helper o auxiliadoras (Th), que promueven las respuestas inmunes, y las células T reguladoras (Treg), que regulan tales respuestas. 9 Algunas células Th son Th1, Th2, y Th17.
Las Th17 desempeñan un papel funcional importante en la acción inflamatoria. 9 Las células T nativas se diferencian en Th17 mediante IL-1β, IL-6, IL-21 y el factor de necrosis tumoral beta (TGF-β). La IL-17, producida por Th17, agiliza la inflamación al actuar sobre células inmunes y activar los osteoclastos por inducción del activador del receptor del ligando factor nuclear kappa B (RANKL) en los fibroblastos sinoviales.
Por otra parte, citoquinas como IFN-γ, IL-4, y CTLA (del inglés, cytotoxic T-lymphocyte-associated protein 4), producidas por Th1, Th2 y Treg, respectivamente, regulan la diferenciación de los osteoclastos. 9 En la AR existe un desbalance en la relación Th17/Treg, porque Th17 se activa mucho más que Treg.
Los fibroblastos sinoviales contribuyen también a la patogenia de la AR, por la liberación de enzimas degradantes de la matriz: metaloproteinasas de la matriz y catepsinas, que provocan destrucción del cartílago. 10 Los osteoclastos se diferencian de las células madre y producen matriz ósea. En su superficie estas células expresan RANKL, esencial en la formación de los osteoclastos. Las células endoteliales expresan moléculas de adhesión que facilitan la fijación y migración a través del endotelio de los leucocitos (fig. 1).
Inflamación
Para entender la fisiopatología de la AR es necesario conocer los mecanismos implicados en la inflamación, respuesta inmune innata y adquirida a diferentes agresiones endógenas (internas) o exógenas (externas). 11 Según el tiempo de evolución, puede ser aguda o crónica.
La inflamación aguda se caracteriza por rubor, calor, dolor, tumor e impotencia funcional, por la acumulación de leucocitos, proteínas plasmáticas y derivados de la sangre en los sitios lesionados.11Aunque los mecanismos de respuesta innata participan en la inflamación crónica, la inmunidad específica mantiene el proceso en el tiempo y causa daño tisular.
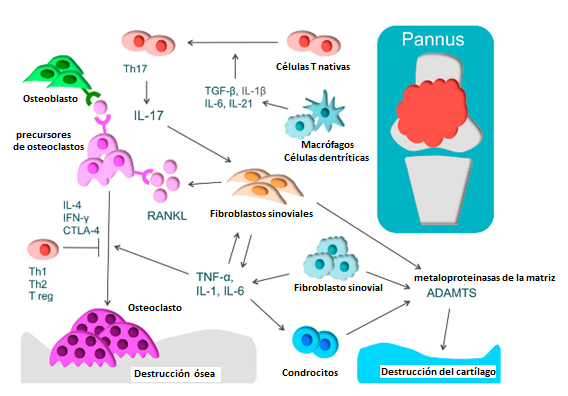
Dentro de los mediadores del proceso inflamatorio se encuentran moléculas y células. 11 Las citoquinas proinflamatorias, como las IL-1, IL-6 e IL-8, el TNF-α y el interferón gamma (IFN-γ), son detectadas en plasma en las primeras 24 horas.
La activación de los macrófagos, que sintetizan el TNF-α en pulmón, riñones e hígado, estimula la producción por los linfocitos, los macrófagos y las células endoteliales de las siguientes citoquinas: IFN-γ, factor estimulante de las colonias de neutrófilos (FECN) y factor activador plaquetario (PAF).
La IL-32 es una citoquina proinflamatoria sintetizada por las NK (del inglés, natural killer), monocitos, células epiteliales y linfocitos T, que participa en la patogenia de la AR. 11 Estimula la producción de la proteína-2 inflamatoria de macrófagos (MIP-2), así como de varias quimioquinas y citoquinas inflamatorias, como son IL-1𝛽, IL-6, IL-8 y TNF- α. IL-32𝛽 (uno de los subtipos), y está implicada en el aumento de la adhesión de células inmunes a las células endoteliales activadas.
Algunos estímulos, como la secreción de IL-1, lipopolisacáridos y TNF-α, pueden aumentar los niveles de leptina y de manera bidireccional esta regula la producción de TNF-α, IL-1 y IL-6, citoquinas proinflamatorias que favorecen la inflamación.
Son muchas las células que participan en el proceso inflamatorio. Las células dendríticas son centinelas en los sitios de intercambio con el medio, como la piel. Son las únicas que viajan a los órganos linfoides secundarios donde presentan péptidos y secretan citoquinas que direccionan la respuesta inmune.11
Los mastocitos de localización perivascular liberan mediadores que actúan como aminas biógenas, quimioquinas y mediadores lipídicos. Entre estas sustancias, cabe mencionar quimasas, histamina, renina, prostaglandinas, leucotrienos y numerosas citoquinas proinflamatorias mencionadas.
Los neutrófilos tienen función secretora, pues al activarse aumentan su degranulación con la liberación de enzimas proteolíticas. Producen radicales libres de oxígeno, que causan la peroxidación de fosfolípidos de la membrana celular y se formen leucotrienos como última estación de la cascada inflamatoria. Una vez contenido el proceso, otras células pueden infiltrar el sitio de lesión como eosinófilos, NK y basófilos.11
En el proceso inflamatorio se favorece la diferenciación de los linfocitos T en dos tipos de poblaciones: Th1 secretores de citoquinas proinflamatorias y los Th2, que producen citoquinas antiinflamatorias, y de esta manera regulan el proceso. 11 Th1 y Th2 amplifican la respuesta inmune adaptativa que permite la acomodación al proceso inflamatorio.
Las Th17, definidas por la secreción selectiva de IL-17, se consideran una línea distinta de las células T auxiliadoras CD4+, que son reguladas por citoquinas Th1 y Th2.12 La IL-17A es la citoquina proinflamatoria producida por Th17, aunque estas células también sintetizan IL-21, IL-22 e IL-26.
En la AR, la IL-17A induce la síntesis de mediadores proinflamatorios desde los fibroblastos sinoviales, macrófagos y condrocitos. Las concentraciones de IL-17 producidas por las células T auxiliadoras CD4+ en la sinovial son significativamente más altas en pacientes con AR que en pacientes con osteoartritis. 12
Patogenia de la artritis reumatoide
Aunque la etiología exacta de la AR no se comprende bien, los avances en las últimas décadas han establecido que es una enfermedad autoinmune inflamatoria crónica, con factores de riesgo genéticos y ambientales. 13
En la patogenia de la AR parecen involucrados tanto el sistema inmune innato como adaptativo. 13 El modelo actual plantea que las interacciones entre células del sistema inmune innato y adaptativo producen inflamación crónica de la sinovial de la articulación, con cambios estructurales y eventual destrucción del cartílago y hueso.
La AR se caracteriza por la hiperplasia de la sinovia, síntesis de citoquinas, quimoquinas, autoanticuerpos, como el factor reumatoideo y anticuerpo contra proteína citrulinada (ACPA, del inglés anticitrullinated protein antibody), osteoclastogénesis, angiogénesis y consecuencias sistémicas, como trastornos cardiovasculares, pulmonares, psicológicos y esqueléticos. 14 Las citoquinas desempeñan un papel crítico en la patogenia de AR.
Citoquinas en la artritis reumatoide
Las citoquinas implicadas en la AR son numerosas y varias se han mencionado: TNF-α, IL-1, IL-6, IL-7, IL-15, IL-17, IL-18, IL-21, IL-23, IL-32, IL-33 y GM-CSF.15
Algunos estudios informan un rol prominente de GM-CSF y en menor medida de IL-3 en las enfermedades autoinmunes como la AR. 16 La deficiencia o neutralización de GM-CSF evita la progresión de la enfermedad en modelos de animales con artritis, mientras su administración exacerba los síntomas.
El GM-CSF, derivado de las células T, incrementa la inflamación crónica, aunque no es indispensable para la iniciación de la artritis. Además, las células del estroma y sinoviales constituyen la principal fuente de esta citoquina; es importante que sus concentraciones en el líquido sinovial se incrementan en pacientes con AR. 16
Los anticuerpos bloqueadores de GM-CSF se están evaluando en ensayos clínicos de pacientes con AR moderada y severa con favorables resultados iniciales. 16
Citoquinas como mediadores de la artritis reumatoide preclínica
Los genes que codifican componentes funcionales del sistema inmune se concentran en regiones cromosómicas asociada con el desarrollo e historia natural de la AR. 4 Las proteínas de estos genes implicados son las citoquinas (IL-2, IL-21, G-CSF y GM-CSF), sus receptores (para IL-6, IL-20, IFN-γ e IL-2) y elementos de la maquinaria de señalización, como TYK2, STAT4 y TNFAIP3.
Ahora se están comprendiendo los mecanismos genéticos y no genéticos por los cuales estas variantes podrían trastornar la homeostasia de las citoquinas para conferir riesgo de enfermedad, lo que es evidente por las concentraciones elevadas de citoquinas circulantes en personas que desarrollarán AR en relación con personas sanas. 4
En personas con predisposición genética, los mediadores o citoquinas pueden elevarse en las articulaciones, médula ósea o en cualquier lugar de la periferia, aunque su presencia refuerza la probable contribución de las citoquinas a la desregulación inmune general y sistémica antes del comienzo de la sinovitis.
Los interferones tipo I, como el prototipo IFN-α, producido principalmente por las células dendríticas plasmocitoides, proporcionan un ejemplo de modulación de autoanticuerpos mediada por citoquinas. 4 Estos factores promueven funciones vinculadas a las enfermedades autoinmunes, como la diferenciación de las células B.
Dentro de los autoanticuerpos detectados en pacientes con AR se encuentran los ACPA de la sinovial, principalmente contra fibrinógeno, filagrina, α-enolasa y vimentina, el colágeno tipo I y II, la actina y las histonas.17,18,19,20
La citrulinación es una modificación postraduccional en residuos de argininas, que forman citrulina. 21 Las proteínas encargadas de la citrulinación son un grupo de enzimas llamadas peptidil arginina desaminasa, de las cuales la tipo 4 se relaciona con excesiva citrulinación de proteínas en la AR. Esta modificación les confiere características de antigenicidad a las proteínas diana, por lo que son reconocidas por células B infiltradas en la articulación, las cuales las fagocitan y producen anticuerpos contra estas proteínas.
Los ACPA se pueden detectar con la prueba de anticuerpos contra péptidos citrulinados cíclicos (anti-CCP) en el 80% de los pacientes con AR, con una especificidad mayor del 98%.18 Los ACPA son anticuerpos de isotipo IgG en su mayoría, aunque también se pueden encontrar isotipos IgA, IgM e IgE. 21 En un estudio en Colombia en pacientes con AR la frecuencia de factor reumatoide y anti-CCP y doble positividad fue: 86, 83 y 80%, respectivamente; y en otras enfermedades reumatológicas: 12,5%; 6% y 1%. 22
ACPA constituye el más específico marcador serológico para el diagnóstico de AR y ha sido incluido por American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) 2010, en los criterios de clasificación de la AR. 17 ACPA también es un predictor de erosión ósea y emerge antes del inicio de la sinovitis durante la fase preclínica de la autoinmunidad. Sin embargo, se requiere determinar el valor real de la reducción de ACPA en pacientes con AR.
Citoquinas en la transición a la cronicidad
Los rasgos distintivos de la sinovitis en la AR son la proliferación de fibroblastos sinoviales, la angiogénesis y el reclutamiento de leucocitos como los linfocitos T y B, monocitos, macrófagos y mastocitos, procesos que originan hipertrofia sinovial y la invasión de cartílago y hueso por las células inflamatorias activadas. 4
Las citoquinas son fundamentales en el origen y desarrollo de las lesiones articulares en la AR. La activación de los osteoclastos del hueso alrededor de las articulaciones conduce a la resorción y se forman las erosiones óseas características de la enfermedad. 3 IL-6 y TNF-α tienen potentes efectos sistémicos que favorecen algunas comorbilidades vistas en AR como trastornos del metabolismo del colesterol y la aterosclerosis. 4
En relación con la inmunidad adaptativa, dos citoquinas inducen la secreción de sPD (del inglés, soluble programmed cell death-1) por las células T CD4+, lo que compromete la regulación mediada por PD-1 de la activación de estas células en pacientes con artritis. 4 Las células T CD4+ reguladoras están disminuidas, lo que contribuye al desequilibrio entre los componentes efectores y reguladores de la inmunidad.3
Las evidencias científicas apuntan a la importancia de las citoquinas en la fase de la enfermedad y las implicaciones en el tratamiento. 4 En la fase preclínica de la enfermedad, se debe considerar también el estado de los autoanticuerpos. Por ejemplo, en pacientes con AR precoz no tratada, las concentraciones elevadas de IL-20 e IL-24 discriminan a las personas seropositivas y predicen la erosión ósea.
La homeostasia inmune se podría restaurar durante la AR. Por ejemplo, la resolución de la sinovitis dependiente de IL-9, en modelos de ratones, sugiere el posible papel regulador de esta citoquina. 4 En pacientes con AR activa, existe una gran cantidad de células linfoides innatas tipo 2 productoras de IL-9 en la circulación y en la sinovia, en comparación con pacientes tratados y controles.
Papel de IL-6
Algunos le atribuyen un papel fundamental en la fisiopatología de la AR a la IL-6, por ser una citoquina que controla múltiples procesos biológicos como la respuesta inmune innata y específica, la inflamación y la hematopoyesis. 23 Las concentraciones elevadas de IL-6 en el líquido sinovial y en suero favorecen la sinovitis y la resorción ósea, lo que incrementa el riesgo de destrucción articular. Se detecta raramente en personas sanas. 24
La IL-6 es una citoquina secretada por los linfocitos T (células T CD4), linfocitos B, monocitos, macrófagos, neutrófilos, células dentríticas, queratinocitos, células endoteliales, fibroblastos, sinoviocitos y células tumorales. 23,24 Diversos estímulos, como otras citoquinas proinflamatorias (TNF-α, IFN-g e IL-1), la IL-32, infecciones virales o traumatismos, desencadenan la producción de IL-6.
Se la considera una citoquina fundamental en el proceso de diferenciación del linfocito T nativo hacia el linfocito Th17 o hacia el Treg. 23 Por lo tanto, la IL-6 tiene un papel determinante en la regulación celular, tanto en las fases tempranas de la AR como en el desarrollo de las manifestaciones sistémicas.
IL-6 activa los neutrófilos y macrófagos y causa que las células Th se diferencien en Th17, además de inhibir las células Treg, con un desbalance Th17/Treg que compromete la tolerancia inmunológica. 24 También promueve la diferenciación de las células B en células productoras de anticuerpos y las células T CD8 en células T citotóxicas.
La unión de la IL-6 a su receptor induce la homodimerización de la glucoproteína (gp) 130, lo que da lugar a la fosforilación de tirosina quinasas de tipo JAK y la posterior activación de los transductores y activadores de señales de la transcripción (STAT) 1 y STAT-3. 23 La expresión del receptor de la IL-6 (IL-6R) de la membrana se limita a los hepatocitos, leucocitos y megacariocitos, y el IL-6R soluble está presente en la circulación periférica y en los sitios de inflamación.
Estudios en ratones sugieren que la activación de la inmunidad de la mucosa intestinal precede el desarrollo de artritis y las células Th17 tienen un papel dependiente de la microbiota en esta enfermedad. 25 Estos autores plantean que la estratificación de pacientes, guiados por la microbiota, podría mejorar las terapias dirigidas a Th17.
Vía Janus quinasa de señalización de citoquinas
La ruta de señalización de las proteínas de la familia Janus quinasa (JAK) está implicada en la patogenia de muchas enfermedades autoinmunitarias, como la AR. 26 Una gran cantidad de citoquinas implicadas en el desarrollo de estas enfermedades utilizan esta vía de señalización para transducir señales intracelulares.
Las proteínas JAK son una familia de tirosina quinasas del citoplasma celular (JAK-1,JAK-2, JAK-3 y tirosine kinase 2:TyK 2) que se expresan ubicuamente en todas las células, excepto en la JAK-3, restringida a células hematopoyéticas. 3 Estas enzimas se asocian a la región citoplasmática de los receptores de citoquinas en forma de dímeros. Cada combinación de JAK y/o TyK es modulada por estímulos específicos y ejerce una función diferente sobre la señalización celular y, por consiguiente, sobre el sistema inmunitario.
Las vías JAK son vías de señalización utilizadas por numerosas citoquinas para regular la respuesta inmunitaria e inflamatoria. Cada proteína JAK muestra especificidad por receptores de citoquinas y cada par de JAK es específico de un grupo diferente de citoquinas. 3 Muchas citoquinas importantes en la patogenia de la AR utilizan las vías JAK.
La unión de las citoquinas a sus receptores activa las proteínas JAK. 3 La activación de las JAK fosforila los factores de trascripción signal transducers and activators of transcription (STAT). Es una familia de 7 miembros (STAT 1, 2, 3, 4, 5a, 5b y 6), que, al ser fosforilados por JAK, se dimerizan y se traslocan al núcleo, donde se unen a los genes diana, activando la transcripción génica. La activación de la vía JAK estimula la producción de citoquinas que contribuyen a mantener la inflamación y provoca daño de las articulaciones.
Diseño de medicamentos
El bloqueo de la señalización de citoquinas proinflamatorias ha revolucionado el tratamiento de los pacientes con AR severa. 4
Tocilizumab, un agente bloqueador del receptor de IL-6, se indica en el tratamiento de enfermedades autoinmunes como AR y artritis idiopática juvenil. 24,25,26,27 Este anticuerpo, primer medicamento exitoso en el bloqueo del receptor de IL-6, fue diseñado por el equipo de Tadamitsu Kishimoto y aprobado por la U.S. Food and Drug Administration (FDA) en 2010 para el tratamiento de AR; posteriormente se aprobó para otras enfermedades. 24
Recientes ensayos clínicos se efectúan para evaluar la eficacia de los nuevos inhibidores de IL-6, como: siltuximab, sirukumab, clazakizumab y olo-kizumab, mientras sarilumab se aprobó para la AR en 2017. 13 A diferencia de tocilizumab, estos agentes se unen a IL-6 y no a su receptor, con la excepción de sarilumab.
Estos conocimientos tienen aplicación práctica también en el diseño y empleo exitoso de inhibidores de TNF-α en pacientes que no responden a los medicamentos tradicionales modificadores de la enfermedad. Los inhibidores de TNF-α aprobados para el tratamiento de la AR son adalimumab, certolizumab pegol, etanercept, golimumab e infliximab. 28,29
El primer agente anti- TNF-α disponible fue infliximab, desarrollado por Jan Vilcek. 13 Es un anticuerpo monoclonal quimérico aprobado para el tratamiento de la AR en 1999, después que los ensayos clínicos iniciales demostraron una mejoría sustancial en los pacientes.
Otro ejemplo es la anakinra, un inhibidor de IL-1 utilizada en la AR. 30 El bloqueo de IL-1 por anakinra evita la unión de IL-1a e IL-1b al receptor y sus efectos inflamatorios, el daño tisular y la disfunción orgánica. 30 Comparado con otros agentes biológicos, este medicamento es bastante seguro porque son raras las infecciones oportunistas.
Las células Th17 juegan un rol en la patogenia de los trastornos autoinmunes como la AR. Modelos en ratones de artritis inducible con sobreexpresión de IL-17 demuestran que esta citoquina contribuye al desarrollo de la artritis, mientras la deleción genética de citoquinas importantes para la diferenciación de Th17 protege contra la artritis. 13
Se prueban agentes inhibidores de IL-17 en la AR: ixekizumab, secukinumab y brodalumab. 13 Ixekizumab y secukinumab son anticuerpos monoclonales humanos que reconocen a IL-17, mientras brodalumab es un anticuerpo monoclonal humano contra el receptor de IL-17.
Se están ensayando fármacos inhibidores de las proteínas JAK. 26 Tofacitinib y baricitinib, los primeros aprobados para el tratamiento de la AR, están en estudio para el tratamiento de otras enfermedades autoinmunitarias. 26,31 Asimismo, otros de estos inhibidores se encuentran en diferentes fases de desarrollo.
Conclusiones
Los trastornos fisiopatológicos en la AR, complejos, multifactoriales, interrelacionados y sometidos a debate científico, parecen vincularse a la síntesis excesiva de citoquinas proinflamatorias producto de las alteraciones del sistema inmunológico, del sistema neuroendocrino y de factores ambientales.
Los avances experimentados en la comprensión de la fisiopatología de la AR y en específico sobre el importante rol de las citoquinas en el origen y desarrollo de esta enfermedad, han permitido el diseño y aplicación de nuevos fármacos con resultados satisfactorios y promisorias expectativas para el futuro.

