










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol v.19 n.2 Ciudad de la Habana Mayo-ago. 2008
PRESENTACIÓN DE CASOS
Feocromocitoma. Presentación de un caso clínico
Pheochromocytoma. A clinical case report
Maité Cabrera GámezI; Silvia Turcios TristáII; Manuel FuentesIII; Teresa González CaleroI; Marelis Yanes QuesadaII; Cossette Díaz SocorroIV
IEspecialista de I Grado en Endocrinología y Medicina General Integral. Instituto Nacional de Endocrinología. La Habana, Cuba.
IIEspecialista I Grado en Endocrinología y Medicina General Integral. Investigadora Agregada. Instituto Nacional de Endocrinología. La Habana, Cuba.
IIIEspecialista de II Grado en Cirugía General. Profesor Consultante. Hospital "Manuel Fajardo". La Habana, Cuba.
IVEspecialista de I Grado en Medicina General Integral y Residente de Endocrinología. Instituto Nacional de Endocrinología. La Habana, Cuba.
RESUMEN
El feocromocitoma es un tumor de células cromafines, que puede localizarse en los territorios derivados de la cresta neural, o en el trayecto que siguen estas células hasta su localización definitiva. Por lo general se ubican en el abdomen (90 % de los casos) particularmente en las glándulas suprarrenales y en el órgano de Zuckerkandl. Pueden causar una amplia variedad de síntomas, dada su capacidad de secretar catecolaminas, particularmente noradrenalina y adrenalina, en cantidades variables e intermitentes. Clínicamente pueden ser asintomáticos o presentarse con hipertensión arterial paroxística, o permanente con o sin paroxismos, acompañados de la triada clásica de cefalea, hiperhidrosis y taquicardia. Teniendo en cuenta lo anterior decidimos presentar un caso clínico de un paciente de sexo masculino, de 40 años de edad, con antecedentes de hipertensión arterial controlada, aparentemente de tipo esencial de varios años de evolución, con el diagnóstico de tumoración suprarrenal derecha incidental, aparentemente no funcionante, y que después de la palpación abdominal pre-quirúrgica, comienza con una crisis paroxística hipertensiva severa. Se prepara con alfa bloqueador y se interviene con informe anatomopatológico de feocromocitoma de suprarrenal derecha.
Palabras clave: Feocromocitoma, hipertensión, tamaño suprarrenal.
ABSTRACT
The pheochromocytoma is a tumor of the chromaffin cells that may be located in territories derived from the neural crest or in the way these cells follow to their definitive localization. Generally, they are located in the abdomen (90 % of the cases), particularly in the suprarenal glands and in Zuckerkandl's organ. They may cause a wide variety of symptoms due to their capacity for secreting catecholamine, specifically noradrenalin and adrenaline in variable and intermittent amounts. Clinically, they may be asymptomatic or appear with paroxistic arterial hypertension, or permanent with or without paroxisms, accompanied with the classic triad of headache, hyperhydrosis and tachycardia. According to the above-mentioned, it was decided to present a clinical case of a 40-year-old male patient with history of apparently essential controlled arterial hypertension of several years of evolution, with the diagnosis of apparently non-functional incidental right suprarenal tumor that after presurgical abdominal palpation began with a paroxistic hypertensive severe crisis. The patient was prepared with an alpha-blocker and he was operated on. A pheochromocytoma of the right suprarenal gland was confirmed in the anatomopathological report.
Key words: Pheochromocytoma, hypertension, suprarenal size.
INTRODUCCIÓN
El feocromocitoma fue descrito por Frankell en 1886 en las suprarrenales de un niño que murió de shock, aunque entonces se clasificó como un angiosarcoma. Neusser asoció por primera vez este tumor con la hipertensión arterial (HTA), y en 1892 Labbé y otros lo relacionaron con la HTA paroxística. El primer diagnóstico clínico de la enfermedad lo realizaron en 1926 Vázquez y Donnelot. El nombre de feocromocitoma se le debe a Pick (1909). El primer tratamiento quirúrgico con éxito lo llevó a cabo Roux en 1926.1
La frecuencia de feocromocitoma en pacientes hipertensos ha sido estimada en menos de un 1 %, sin embargo en el 0,1 % las autopsias se descubre la presencia de un feocromocitoma, lo que demuestra que dicha incidencia puede estar infravalorada.2
Los feocromocitomas son tumores del sistema nervioso simpático que se desarrollan a partir de las células cromafines, y que se caracterizan por la producción excesiva de catecolaminas. En el 85 % de los casos se localizan en la médula suprarrenal y el 15 % restante son extrasuprarrenales (paragangliomas, el 90 % son esporádicos y el 10 % pueden formar parte de una enfermedad neoplásica endocrina múltiple [MEN] tipo 2A o 2B, o bien asociarse a otros síndromes neuroectodérmicos).3
Son tumores total o parcialmente encapsulados, muy vascularizados, y en general, mayores de 3 cm de diámetro y con un peso medio de 100 g, aunque se han registrado variaciones desde 1 g hasta casi 4. Al corte, la superficie tiene un color gris pálido o pardo, y pueden observarse áreas de hemorragias, necrosis o formación de quistes, en especial en las lesiones de mayor tamaño. Histológicamente son de células pleomórficas de gran tamaño con citoplasma basófilo o eosinófilo, dispuestas en nidos, y muestran atipias nucleares y mitosis, con frecuencia sin expresión de malignidad. Debido a que los feocromocitomas malignos y benignos pueden tener un aspecto histológico idéntico, el único criterio absoluto de malignidad es la enfermedad metastásica (ganglios linfáticos, hígado, pulmón y hueso).4
Con el objetivo de destacar la importancia que continúa teniendo en la práctica clínica el interrogatorio y el examen físico minucioso, a pesar de los novedosos medios diagnósticos con que contamos hoy, y teniendo en cuenta el subdiagnóstico de esta causa curable de HTA, nos motivamos a presentar el caso siguiente.
PRESENTACIÓN DEL CASO
Paciente de sexo masculino, de 40 años de edad, mestizo, con antecedentes personales de HTA aparentemente esencial, medicado con captopril (25 mg cada 8 h) y clorodiazepóxido (10 mg cada 8 h), bien controlado. Seis meses antes de su ingreso en el Instituto Nacional de Endocrinología fue intervenido quirúrgicamente por hemotórax izquierdo postraumático, y a partir de este momento comenzó a notar molestias crónicas lumboabdominales contralaterales y cambios en el hábito intestinal (constipación). Por la persistencia de la sintomatología referida le indicaron estudios ultrasonográficos, en los que se detecta una imagen tumoral en territorio de la suprarrenal derecha.
Hábitos tóxicos: consumo de cigarrillos (22 años de fumador).
Alergia a medicamentos: penicilina, tetraciclina, aspirina y cotrimoxazol.
Datos positivos al examen físico durante el ingreso en nuestro servicio:
- Peso: 79 kg
- Talla: 173 cm
- IMC: 26,3 kg/m2 (sobrepeso).
- TA: 140/100 mmHg. En ocasiones presentó cifras elevadas de tensión arterial que se controlaron con antihipertensivos (captopril y nifedipino).
Resumen de los exámenes (tabla).

US abdominal: masa ecogénica heterogénea que mide 69 x 72 mm en territorio de la suprarrenal derecha. No alteraciones en el resto de los órganos del abdomen.
Rx de tórax: índice cardiotorácico normal, no lesiones pleuropulmonares.
TAC abdominal (19/4/06): se realiza examen simple, en el que se observa una masa sólida (44 UH) en proyección de la glándula suprarrenal derecha que mide 5 x 8 cm. No hay alteraciones en el territorio de la suprarrenal izquierda. Se realiza examen endovenoso (EV) en el que se observa glándula suprarrenal derecha tumoral, que mide 78 x 60 mm, que capta contraste, de 58 UH. No hay otras alteraciones.
Por las características imagenológicas (tamaño > de 6 cm, densidad de unidades Hounsfield mayor de 20) se decide indicar tratamiento quirúrgico, y teniendo en cuenta los APP de HTA del paciente se comienza su preparación quirúrgica con alfa bloqueadores (terazosin a la dosis de 5 mg/día). Al cuarto día de preparación, y luego de una maniobra de exploración abdominal profunda realizada en otras ocasiones durante su ingreso, comienza con un cuadro súbito de HTA severa (220/160 mmHg), acompañada de sensación de calor, rubor facial, opresión torácica, sudoración, taquicardia (160 latidos/min), cefalea universal, intensa, pulsátil; con cuadros de hipotensión súbita (50/30 mmHg) y bradicardia, que duró aproximadamente 1 h, con sensación referida de muerte. Se trasladó a la Unidad de Cuidados Intensivos, donde mantuvo episodios similares, con una frecuencia de 10 a 20 veces al día en las primeras 48 h, momento a partir del cual comienza a mejorar. Se realiza intervención quirúrgica, con el diagnóstico clínico e imagenológico de un feocromocitoma, sin complicaciones hemodinámicas significativas.
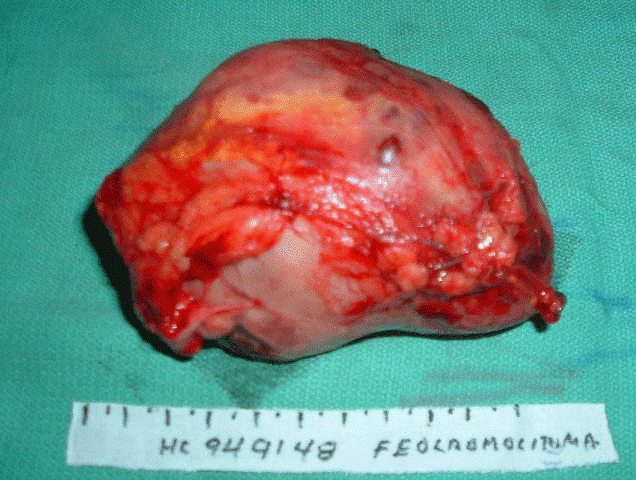
Informe de anatomía patológica: se recibe masa de tejido ovalada que mide 11 x 7 x 6 cm y pesa 130 g, superficie pardo amarillenta, tejido pardo grisáceo, con extensas áreas de hemorragias y otras de necrosis con muy escasas áreas viables, de las que se pesan 5 fragmentos.
Nota: el tumor muestra marcadas áreas de necrosis, hemorragia y atipicidad celular. Se recomienda evolución estricta.
DISCUSIÓN
Las catecolaminas se forman a partir del aminoácido tirosina por un proceso de hidroxilación y de descarboxilación. Sus acciones biológicas sobre el metabolismo, y en particular sobre el aparato cardiovascular, son variadas y están mediados por receptores específicos para catecolaminas. La elevación de sus concentraciones plasmáticas aumenta la tensión arterial por diferentes mecanismos: al incrementar el gasto cardiaco y la resistencia vascular periférica por la acción vasoconstrictora en las arteriolas, además induce la liberación de renina a partir del riñón, lo que provoca secundariamente el aumento en la producción de angiotensina II.5 La acentuación de los síntomas del feocromocitoma se debe al bloqueo en la recaptación de noradrenalina desde las terminales nerviosas, que ocasiona una marcada elevación de los niveles plasmáticos de noradrenalina.6
La clínica puede ser variada, y no guarda una clara relación con el tamaño, localización o aspecto histológico del tumor. Los síntomas más comunes son: cefalea, diaforesis y palpitaciones. Las manifestaciones clínicas pueden ser paroxísticas en un por ciento de los casos. La hipotensión ortostática aparece en menos del 50 % de ellos.7
También pueden tener poca expresividad clínica o ser asintomáticos, circunstancia que ocurre en un 20 % de los casos aproximadamente, sin embargo esta característica pudiera corresponder con un subregisto de casos, teniendo en cuenta que en algunos reportes se cita que más del 50 % de los feocromocitomas se descubren en el examen autópsico. Se ha estimado que un 5-10 % de los incidentalomas son feocromocitomas, y esto se debe también a sus manifestaciones clínicas silentes durante años, y se describe que una situación de estrés puede precipitar una crisis hipertensiva.8
Los síntomas también pueden precipitarse con el ejercicio, la maniobra de Valsalva, el coito, la defecación, la micción (feocromocitoma vesical), la inducción anestésica, la cirugía, el parto, o bien tras la administración de fármacos, tales como: b-bloqueantes, antidepresivos tricíclicos, fenotiacinas, metoclopramida, glucagón y fármacos citotóxicos, entre otros.3 En nuestro paciente, la palpación abdominal se convirtió en el factor precipitante de la crisis paroxística, al estimular la liberación de catecolaminas, que se expresó clínicamente por los cambios hemodinámicos ya mencionados, y que fueron de gran severidad, con peligro para la vida del paciente. La localización del tumor en el caso presentado se correspondió con lo reportado en la literatura como de mayor frecuencia (derecha);9 pero esto no sucedió con sus dimensiones, que se señala se encuentran por lo general entre 3 y 5 cm de diámetro,10 y el de nuestro caso las superó significativamente (figura).
CONCLUSIONES
No obstante la poca frecuencia diagnóstica del feocromocitoma, debemos tenerlo en cuenta en el diagnóstico diferencial de toda masa tumoral suprarrenal, a pesar de tener el criterio de ser un incidentaloma. Ser cautelosos en la exploración física, y tener en cuenta toda la historia patológica anterior referida, pudiera ser de utilidad en la definición de una conducta, garantizando un menor riesgo para la vida del paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Méndez SA. Adrenales. En: Breve historia de la endocrinología. La Habana: Editorial Científico Técnica; 1975.p.131-8.
2. Norton JA. Adrenal tumors. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds.: Cancer: Principles and Practice of Oncology. 7th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.p.1528-39.
3. Lairmore TC, Ball DW, Baylin SB. Management of pheochromocytomas in patients with multiple endocrine neoplasia type 2 syndromes. Ann Surg. 217(6):595-601.
4. Robbins SL, Kumar V, Cotran RS. Patología estructural y funcional. Madrid: Mc Graw-Hill-Interamericana de España; 1998.
5. Lavin N. Endocrinología y metabolismo. Madrid: Marban Libros, S.L; 2003.p.149-53.
6. Dluhy R, Lawrence J, Williams RH. Hipertensión de origen endocrino. En: Tratado de Endocrinología Clínica. RH Williams. 10ma ed. USA: Ed. Elservier; 2000.p.677.
7. Munakata M, Aihara A, Imai Y, Noshiro T, Ito S, Yoshinaga K. Altered sympathetic and vagal modulations of the cardiovascular system in patients with pheochromocytoma: their relations to orthostatic hypotension. Am J Hypertens. 1999;12:572-80.
8. Mateo A, Rentería M. Feocromocitoma: revisión u manejo quirúrgico. Rev Hosp Gral Dr. M Gea González. 2000;3(4):170-81.
9. Amberson JB, Vaughan ED, Gray GF. Flow cytometric determination of nuclear DNA content in benign adrenal pheochromocytomas. Urology. 1987;30(2):102-4.
10. Adrenocortical Carcinomas and Adrenal Pheochromocytomas. Mass and Enhancement loss. Evaluation at delayed contrast-enhanced CT. Radiology R.S.N.A. (The Radiological Society of North America. 2005.p.479-85).
Recibido: 12 de mayo de 2008.
Aprobado: 8 de junio de 2008.
Maité Cabrera Gámez. Instituto Nacional de Endocrinología. Calle Zapata esquina C, Vedado, municipio Plaza, Ciudad de La Habana, Cuba. E mail: maite.gamez@infomed.sld.cu

