










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol v.21 n.3 Ciudad de la Habana sep.-dic. 2010
REVISIÓN BIBLIOGRÁFICA
Metilación y expresión de genes en el cáncer diferenciado de tiroides
Methylation and expression of genes in thyroid differentiated cancer
María Teresa Marrero Rodríguez
Licenciada en Biología. Investigadora Agregada. Instituto Nacional de Endocrinología. La Habana, Cuba.
RESUMEN
En los últimos años ha cobrado importancia el estudio de las alteraciones epigenéticas en el desarrollo del cáncer. La metilación del ácido desoxirribonucleico es el cambio epigenético más frecuente e importante hasta ahora estudiado, y tiene un importante papel en la regulación transcripcional de genes. Recientemente se ha observado que existen patrones de metilación anormales en muchos tipos de cánceres, incluyendo el cáncer de tiroides, los cuales conducen a la inactivación de genes supresores de tumores y a la inestabilidad del genoma. La metilación de genes específicos, tales como, el cotransportador de yodo/sodio, la tiroglobulina y el receptor de la hormona estimulante del tiroides en el cáncer diferenciado de tiroides, es una de las causas de fallo en el tratamiento de los pacientes con esta enfermedad. Se ha iniciado el tratamiento con agentes desmetilantes en los pacientes con cáncer de tiroides que presentan una alteración genética por metilación, a fin de corregir estas alteraciones, restablecer la función, y con ello, la posibilidad de que el tratamiento sea efectivo.
Palabras clave: Epigenética, metilación, agentes desmetilantes, ácido desoxirribonucleico (ADN), genes, cáncer diferenciado de tiroides.
ABSTRACT
In past years the study of epigenetic alterations in the cancer development becomes significance. The methylation of desoxyribonucleic acid is the more frequent and important epigenetic change until now studied and play a significant role in the transcription regulation of genes. Recently it was noted the existence of abnormal methylation patterns in many types of cancer, including the thyroid one, which leading to inactivation of tumor suppressors genes and to genome instability. The methylation of specific genes such as the co-transporter of iodine/sodium, the thyroglobulin and the receptor of thyroid stimulant hormone (TSH) in the thyroid differentiated cancer, is one of the failure cause in treatment of patients presenting this disease. In patients with thyroid cancer it has been initiated a treatment with demethylation agents in patients with abovementioned cancer with a genetic alteration due to methylation to correct these alterations, to restore the function and thus the possibility of a effective treatment.
Key words: Epigenetic, methylation, demethylation agents, desoxyribonucleic acid (DNA), genes, thyroid differentiated cancer.
INTRODUCCIÓN
Las células somáticas de un organismo pluricelular tienen básicamente la misma información genética, no obstante, cada uno de los tipos celulares que forman parte del organismo tienen una estructura y función característica. Esto se debe a la expresión diferencial del genoma, la cual es regulada principalmente por mecanismos epigenéticos.
El término epigenética ha sido definido como: "los cambios heredables en la expresión génica que ocurren sin una alteración en la secuencia de nucleótidos del ácido desoxirribonucleico (ADN)".1-4 Así, un mecanismo epigenético puede ser entendido como un sistema complejo para utilizar selectivamente la información genética, activando y desactivando diversos genes funcionales.1-6 Dentro de los procesos más estudiados en este campo, se encuentran la metilación del ADN y la acetilación de las histonas.4
La metilación del ADN constituye un marcador epigenético que identifica la cadena molde durante la replicación del ADN y regula la expresión génica, la impronta genómica y a los transposones.7 Las alteraciones en la metilación del ADN se asocian con diferentes enfermedades, especialmente las relacionadas con afectaciones en el proceso de transformación celular. En este sentido, diversas evidencias han demostrado la importancia de los mecanismos epigenéticos en la regulación transcripcional de genes supresores de tumores y oncogenes.
DESARROLLO
Los cambios en el estado de metilación (hipometilación e hipermetilación) de genes que participan en la reparación del ADN, regulación del ciclo celular y crecimiento y adhesión celular, promueven la transformación celular maligna. Dichas alteraciones pueden aparecer antes en células premalignas o durante la progresión del tumor, y participar en la severidad y/o en el grado de malignidad. En la actualidad se han logrado detectar alteraciones epigenéticas en varios tipos de cánceres, como el de colon, estómago, próstata, mama y tiroides. En este último, se ha observado metilación en la región promotora de los genes que codifican para proteínas específicas del tiroides, como son: el receptor de tirotropina (TSHr) y el cotransportador yodo/sodio (NIS),5 que conducen a un silenciamiento de los mismos genes, y por tanto, a una progresiva pérdida de la expresión de estas moléculas, lo cual provoca que este tema sea un tópico de gran relevancia médica.
Esta revisión se centra en describir los mecanismos generales de la metilación del ADN y su relación con la expresión de genes en el cáncer diferenciado de tiroides.
1. Metilación del ADN
La metilación es un fenómeno fisiológico importante en la regulación de la expresión de los genes en mamíferos, principalmente durante la embriogénesis, y es de vital importancia para mantener el silenciamiento genético con el fin de regular, de manera adecuada, la expresión de los genes, y asegurar un desarrollo normal del ser humano. Se ha observado en diversas especies de bacterias, algunos hongos, plantas y organismos superiores.
En las bacterias, es parte de un mecanismo de defensa para reducir la cantidad de transferencia génica horizontal entre las especies.6 En los mamíferos, la metilación del ADN se refiere a la inserción de un grupo metilo (CH3) en la posición 5 de la base nitrogenada dioxicitosina (dc) para formar 5-metilcitosina. Es una reacción enzimática catalizada por la ADN metiltransferasa (DNMT) en presencia de un sustrato (S-adenosilmetionina) donador de grupos metilo (figura 1).
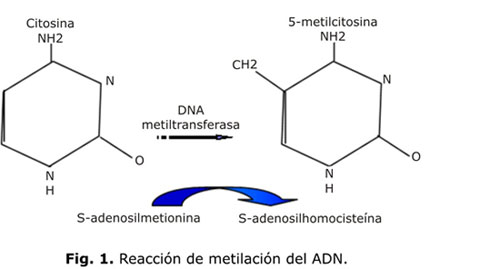
La 5-metilcitosina se encuentra en los dinucleótidos citocina-guanina (CpG), que no están distribuidos uniformemente en el genoma humano. En el 98 % del genoma, los CpG están presentes como promedio 1 vez por cada 80 dinucleótidos, y existen regiones de 200 pares de bases (pb) con una frecuencia 5 veces mayor de dinucleótidos CpG, denominadas islas CpG.8
Aproximadamente del 60 al 90 % de todas las secuencias CpG dispersas en el genoma están metiladas, mientras que las correspondientes a las islas CpG localizadas en la mayoría de los genes de mantenimiento celular, no lo están. En general, las islas CpG se localizan entre la región central del promotor y en el sitio de inicio de la transcripción, observándose represión en la expresión del gen cuando se encuentran metiladas.9
La asociación de la metilación del ADN con la represión génica fisiológica se sugirió por primera vez hace casi 30 años;7 mientras que su participación en procesos patológicos se demostró en 1990, cuando se evidenció la asociación entre la metilación de las islas CpG10 y la inactivación de genes en líneas celulares de origen tumoral.
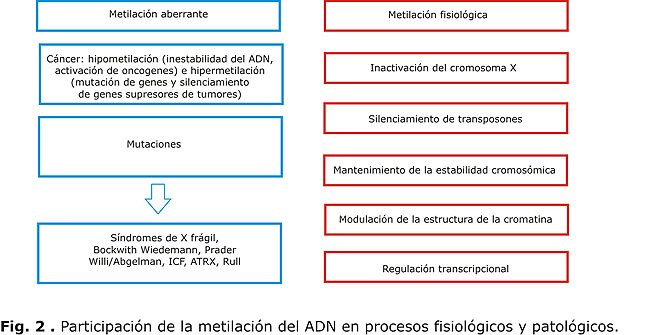
Dentro de los procesos fisiológicos involucrados se encuentran: la regulación transcripcional, el mantenimiento de la estabilidad cromosómica, la modulación de la estructura de la cromatina, la inactivación de uno de los cromosomas x en mujeres, la impronta genómica, y el silenciamiento de retrotransposones.11,12 Por ejemplo, en el caso de la impronta, la metilación de ADN como patrón hereditario, puede determinar cuál de los alelos (el proveniente de la madre o el del padre) se expresa, o si finalmente lo hacen ambos.13,14 La expresión de un solo alelo supondría una menor actividad de la proteína relacionada, mientras que la expresión de ambos, una actividad aumentada, lo que lleva a manifestaciones fenotípicas diferentes que podrían ser patológicas.
Estas variaciones en la impronta podrían explicar las características fenotípicas que desaparecen y reaparecen al cabo de varias generaciones, también aclararía las diferencias registradas en algunas enfermedades como la DM 2, que se manifiesta en diferentes edades en los miembros de una misma familia; e igualmente explicaría enfermedades en gemelos idénticos, como la esquizofrenia, que puede afectar a solo uno de ellos.15,16
Otras enfermedades asociadas con procesos de metilación de ADN son: el immunodeficiency, centromere instability, facial anormalies (síndrome ICF) causado por una mutación que inactiva el gen de la ADN metiltransferasa 3; el síndrome de Rett, causado por mutaciones en el gen MECP2, las cuales definen el espectro de sus formas clínicas del mismo;17 y los síndromes de Prader-Willi y Angelman, causados por la misma deleción en el cromosoma 15, pero con manifestaciones diferentes en dependencia de si el cromosoma afectado es el materno o el paterno por el fenómeno de impronta.18
Se ha encontrado que el ADN de células normales puede ser metilado de novo y propiciar el silenciamiento de genes supresores de tumores, así como la desregulación de los mecanismos de control en el ciclo celular.19,20 Un número significativo de islas CpG son susceptibles de metilación progresiva en ciertos tejidos durante el proceso de envejecimiento o en los procesos neoplásicos; sin embargo, la velocidad con que ocurren estos cambios parece ser muy lenta.21
Recientemente se ha observado que existen patrones de metilación anormales o una metilación aberrante en muchos tipos de cáncer (hipermetilación e hipometilación), los cuales conducen principalmente a la inactivación de genes supresores de tumores y a la inestabilidad del genoma.22 En el caso del cáncer, la investigación se encuentra ante una paradoja: por un lado, se ha demostrado que hay una hipermetilación restringida a determinadas regiones del ADN; y por otro lado, existe una hipometilación generalizada del genoma (figura 2) en presencia de un aumento de la actividad de DNMT.23
La hipermetilación de las islas CpG ha sido el fenómeno más estudiado de los asociados con el cáncer. La hipermetilación de promotores de genes supresores de tumores como p6/CDKN2, Rb, p21, BRCA1, genes de reparación de ADN como hMLH1 y otros, como el receptor de estrógeno, provocan su represión en tejidos cancerosos.24 Se ha propuesto que la hipometilación generalizada podría desestabilizar el genoma, al activar las secuencias transponibles de ADN, denominadas genes saltarines. El descontrol de estos elementos puede causar mutaciones de tipo insercional e inactivar genes de control de la proliferación y el crecimiento.12,25 La hipometilación podría generar una inestabilidad cromosómica, como la observada en el síndrome ICF.12
1.1 Metilación y cáncer
El proceso de carcinogénesis comprende una serie de alteraciones genéticas y epigenéticas que son acumuladas en las células y que terminan por permitir su crecimiento no regulado. Posiblemente el cáncer es uno de los procesos patológicos en el que más se ha estudiado la metilación del ADN. Existen numerosos datos que indican que las células tumorales, invariablemente, presentan diversas alteraciones en el ADN.26
En el origen y desarrollo de células neoplásicas malignas juegan un papel fundamental 2 tipos de genes: los oncogenes y los genes supresores de tumores, cuya activación o inactivación está directamente relacionada con la transformación celular. La gran mayoría de ellos forman parte de rutas de señalización encargadas de controlar el ciclo celular, la apoptosis, la diferenciación celular, la integridad del genoma y las reacciones morfogenéticas.27 Otros factores implicados en el desarrollo del cáncer son los cambios epigenéticos: metilación del ADN y modificación de histonas. De hecho, los cambios en el patrón de metilación pueden ser tan importantes como las mutaciones génicas o el cambio del número de copias de un gen.28
Los patrones de metilación varían en respuesta a cambios en la dieta, polimorfismos genéticos y la exposición a agentes químicos, que, junto con la modificación de histonas, pueden conllevar a daños severos a la salud, como son, las enfermedades congénitas, las neurodegenerativas, la ateroesclerosis y el cáncer.29
Los genomas de las células preneoplásicas, cancerosas y envejecidas comparten 3 cambios importantes en los niveles de metilación, como eventos tempranos en el desarrollo de algunos tumores. Primero, la hipometilación de la heterocromatina, que conduce a una inestabilidad genómica e incrementa los eventos de recombinación mitótica; la segunda, la hipermetilación de genes individuales; y, finalmente, la hipermetilación de las islas CpG de genes constitutivos y genes tumor supresor.
Los 2 niveles de metilación pueden presentarse en forma individual o simultánea. En general, la hipermetilación está involucrada con el silenciamiento de genes y provoca represión transcripcional debido a un cambio estructural de la cromatina, que la hace inaccesible a los factores de transcripción. De esta manera, se inactivan genes supresores de tumores, lo que se pensaba que ocurriese exclusivamente por deleción alélica o mutación, y la hipometilación con la sobre-expresión de ciertas proteínas involucradas en los procesos de invasión y metástasis (oncogenes), que provocan inestabilidad genómica.30
2. Metilación y cáncer diferenciado de tiroides, estrategias actuales para su tratamiento
El cáncer diferenciado de células foliculares del tiroides es la neoplasia endocrina más frecuente, y constituye el 1 % de todos los cánceres en humanos.1 Es generalmente sensible al tratamiento convencional y tiene un buen pronóstico, sin embargo en el 30 % de los pacientes con enfermedad persistente o recurrente, puede ocurrir desdiferenciación celular, caracterizada por la pérdida de las propiedades y funciones específicas de la célula folicular tiroidea, lo que hace que el tumor sea inaccesible a los tratamientos convencionales.
Los tirocitos son células altamente diferenciadas y expresan genes que codifican para proteínas específicas como la tiroglobulina (Tg), tiroperóxidasa (TPO), receptor de tirotropina (TSHr) y el NIS, así como factores transcripcionales, tales como, factor de transformación tiroideo 1 y 2 (TTF-1 y TTF-2) y Pax 8.
Los tirocitos normales, por la presencia de cambios en determinados genes, pueden transformarse en células tumorales y sin embargo conservar propiedades específicas. Una acumulación de alteraciones en genes relacionados con la proliferación y diferenciación celular, puede conllevar al desarrollo progresivo de carcinoma pobremente diferenciado, y por último, a carcinoma indiferenciado. Durante dicho proceso de desdiferenciación, la célula tumoral pierde la capacidad de expresar genes específicos del tiroides, incluyendo aquellos involucrados en la captación y organificación del yodo, síntesis de Tg y expresión del TSHr.31-34
De lo anterior se comprende que la ablación con yodo radiactivo y el tratamiento supresivo resultan inefectivos en este grupo de pacientes. Por otra parte, estos tumores habitualmente no responden a tratamientos alternativos oncológicos (radioterapia externa o quimioterapia sistémica), y aunque el mecanismo molecular del silenciamiento de estos genes es aún desconocido, sí se conoce que la metilación juega un papel importante.
En los tumores tiroideos, muchos de los genes específicos se encuentran metilados en su región promotora, incluyendo los que codifican para el NIS32,35 TSHr.36 El silenciamiento de estos genes puede provocar progresión o desdiferenciación del cáncer de tiroides. Por ejemplo, empleando líneas celulares de tumores tiroideos humanos, se ha encontrado una metilación aberrante en el gen NIS y el consiguiente silenciamiento de su expresión, lo que fue revertido después del tratamiento de las células con agentes desmetilantes.32,37 También se ha reportado que la expresión del gen TSHr se encuentra frecuentemente silenciada en el cáncer epitelial tiroideo asociado con una disminución o ausencia de la captación de yodo regulado por la TSH. Se plantea que una de las vías del silenciamiento de este gen sea la metilación aberrante en su región promotora, demostrada en líneas celulares de tumor tiroideo y muestras de tejido de cáncer de tiroides. Estos resultados confirman los estudios realizados en líneas celulares en ratas38 y muestran que la pérdida de la expresión del gen TSHr es solamente observada en tumores tiroideos malignos.
Dentro de las actuales estrategias para el tratamiento del cáncer folicular del tiroides, la ablación posquirúrgica del remanente tiroideo con yodo radiactivo desempeña un papel fundamental. Esta terapéutica presenta la ventaja de ser también eficaz frente a las metástasis; no obstante, en algunos casos, la célula folicular neoplásica pierde la capacidad para captar y concentrar yodo, y con ello, la posibilidad de ser tratada eficazmente mediante este procedimiento.39,40
La expresión del NIS es necesaria para el transporte del yodo al interior de la célula tiroidea, y por tanto, para la captación de yodo radioactivo durante el tratamiento de las células tumorales. Existen evidencias que algunas líneas celulares de carcinoma papilar presentan modificaciones (cambios epigenéticos, ya sean metilaciones o acetilación de histonas) en el promotor del NIS, lo cual condiciona la disminución de su expresión.40 A diferencia de los cambios genéticos (por ejemplo, las mutaciones) la reversibilidad de los cambios epigenéticos ha conducido a una ampliación de las terapias epigenéticas, seleccionando deacetilasas de histonas (HDAC) y agentes desmetilantes del ADN.41-43
Es bien sabido que ciertos agentes desmetilantes tienen un efecto terapéutico en algunos tipos de cánceres. La 5-azacitidina es un agente desmetilante que ha sido capaz de restaurar la captación de yodo en algunas líneas celulares de cáncer de tiroides.32 Dicha observación se encuentra avalada por el hecho de que el tratamiento de líneas celulares de carcinoma indiferenciado de tiroides logró restablecer la función del NIS.44 Además, dicho tratamiento aumentó la expresión de otras proteínas específicas del tiroides, y logró hacer al tumor más susceptible a la terapia convencional con yodo radiactivo.45
Dichos fármacos logran que pueda iniciarse la transcripción de los genes que estaban relacionados con los nucleosomas afectados, lo que lleva a la diferenciación celular.45 Todo ello apunta a la posibilidad que en el proceso de indiferenciación del cáncer de tiroides también intervengan factores epigenéticos. Es evidente que se necesitan estudios in vivo que avalen el empleo de estos nuevos compuestos que parecen diferenciar de nuevo la célula tumoral tiroidea in vitro, y que muy probablemente la combinación de estas estrategias mejore el efecto terapéutico en este tipo de pacientes que ha perdido la capacidad de concentrar yodo.
Se puede concluir señalando que el 90 % de los tumores tiene una alteración epigenética similar, que impide que los genes inhiban el surgimiento y progresión del cáncer. El conocimiento de los patrones de metilación en las diferentes regiones del genoma permitirá establecer, en forma precisa, la aparición de cambios asociados con el estado de malignidad de diversos tumores. Además, debido a que los cambios epigenéticos son reversibles, el diseño de estrategias terapéuticas encaminadas a corregir las alteraciones en la metilación del ADN —principalmente mediante el uso de agentes desmetilantes— es una ruta importante para mejorar la atención y pronóstico de los pacientes con padecimientos que involucran estas alteraciones, especialmente en aquellos con cáncer de tiroides que pierden la capacidad para captar y concentrar yodo, y con ello la posibilidad de que el tratamiento resulte efectivo.
REFERENCIAS BIBLIOGRÁFICAS
1. Allegrucci C, Thurston A, Lucas E, Young L. Epigenetics and the germline. Reproduction. 2005;129:137-49.
2. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604-9.
3. Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345-50.
4. Laird PW. Cancer epigenetics. Hum Mol Genet. 2005;14 Suppl 1:R65-76.
5. Xing M. Gene methylation in thyroid tumorigenesis. Endocrinology. 2007;148:948-53.
6. Kondo T, Asa SL, Ezzat S. Epigenetic dysregulation in thyroid neoplasia. Endocrinol Metab Clin North Am. 2008;37:389-400.
7. Antequera F, Boyes J, Bird A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 1990;62:503-14.
8. Wen W Ma, Adjei Alex A. Novel Agents on the Horizon for Cancer. Therapy Cancer J Clin. 2009;59:111-37.
9. Costello JF, Plass C. Methylation matters. J Med Genet. 2001;38:285-303.
10. Robertson KD. DNA methylation and chromatin-unraveling the tangled web. Oncogene. 2002;21:5361-79.
11. Strathdee G, Brown R. Aberrant DNA methylation in cancer: potential clinical interventions. Exp Rev Mol Med. 2002;4:1-17.
12. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6-21.
13. Bartolomei MS, Tilghman SM. Genomic imprinting in mammals. Annu Rev Genet. 1997;31:493-525.
14. Abdolmaleky HM, Smith CL, Faraone SV, Shafa R, Stone W, Glatt SJ, et al. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet. 2004;127B:51-9.
15. Chen Y, Sharma RP, Costa RH, Costa E, Grayson DR. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 2002;30:2930-9.
16. Nicholls RD, Saito S, Horsthmke B. Imprinting in Prader-Willi and Angelman syndromes. Trends Genet. 1998;14:194-200.
17. Cheadle JP, Gill H, Flemming N, Maylanrd J, Kerr A, Leonard H, et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Hum Mol Genet. 2000;9:1119-29.
18. Holliday, R. DNA methylation and epigenotypes. Biokhimiya. 2005;70(5):612-7.
19. Mund C, Beier V, Berewunge P, Dahms M, Lyko F, Hoheisel JD. Array-based analysis of genomic DNA methylation patterns of the tumor suppressor gene p16INK4A promoter in colon carcinoma cell lines. Nucleic Acids Research. 2005;33(8):73.
20. Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089-2593.
21. Bird A, Wolffe AP. Methylation induces repression-belts, braces and chromatin. Cell. 1999;99:451-4.
22. Herman JG, Baylin SB. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N Engl J Med. 2003;349:2042-54.
23. Leone G, Teofili L, Voso MT, Lubbert M. DNA methylation and demethylating drugs in myelodysplastic syndromes and secondary leukemias. Haematologica. 2002;87:1324-41.
24. Yuan Y, Mendez R, Sahin A, Dai JL. Hypermethylation Leads to Silencing of the SYK Gene in Human Breast Cancer. Cancer Res. 2001;61:5558-61.
25. Hanahan W. The hallmarks of cancer. Cell. 2000;100:57-70.
26. Kopnin BP. Targets of oncogenes and tumor suppressors: key for understanding basic mechanisms of carcinogenesis. Biochemistry (Mosc). 2000;65:2-27.
27. Jones P, Ay Baylyn SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415-28.
28. Rodenhiser D, Mann M. Epigenetics and human disease: translating basic bilogy into clinical application. CMAJ. 2006;174:341-8.
29. Mesa-Cornejo VM, Barros-Nunez P, Medina-Lozano C. Metilación del ADN: marcador diagnóstico y pronóstico de cáncer. Gac Méd Méx. 2006;142(1):81-2.
30. Mack GS. Epigenetic cancer therapy makes headway. J Natl Cancer Inst. 2006;98:1443-4.
31. Venkataraman GM, Yatin M, Marcinek R, Ain KB. Restoration of iodide uptake in dedifferentiated thyroid carcinoma: relationship to human Na+/I- symporter gene methylation status. J Clin Endocrinol Metab. 1999;84:2449-57.
32. Ringel MD, Anderson J, Souza SL, Burch HB, Tambascia M, Shriver CD, et al. Expression of the sodium iodide symporter and thyroglobulin genes are reduced in papillary thyroid cancer. Mod Pathol. 2001;14:289-96.
33. Arturi F, Russo D, Bidart JM, Scarpelli D, Schlumberger M, Filetti S. Expression pattern of the pendrin and sodium/iodide symporter genes in human thyroid carcinoma cell lines and human thyroid tumors. Eur J Endocrinol. 2001;145:129-35.
34. Neumann S, Schuchardt K, Reske A, Reske A, Emmrich P, Paschke R. Lack of correlation for sodium iodide symporter mRNA and protein expression and analysis of sodium iodide symporter promoter methylation in benign cold thyroid nodules. Thyroid. 2004;14:99-111.
35. Xing M, Usadel H, Cohen Y, Tokumaru Y, Guo Z, Westra WB, et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: a marker of malignancy and a cause of gene silencing. Cancer Res. 2003;63:2316-21.
36. Berlingieri MT, Musti AM, Avvedimento VE, Di Lauro R, Di Fiore PP, Fusco A. The block of thyroglobulin synthesis, which occurs upon transformation of rat thyroid epithelial cells, is at the transcriptional level and it is associated with methylation of the 5' flanking region of the gene. Exp Cell Res. 1989;183:277-83.
37. Ringel MD, Anderson J, Souza SL, Burch HB, Tambascia M, Shriver CD, et al. Expression of the sodium iodide symporter and thyroglobulin genes are reduced in papillary thyroid cancer. Mod Pathol. 2001;14:289-96.
38. Basolo F, Pisaturo F, Pollina LE, Fontanini G, Elisei R, Molinaro E, et al. N-ras mutation in poorly differentiated thyroid carcinomas: correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid. 2000;10:19-23.
39. Carrasco N. The thyroid sodium-iodide symporter (NIS): cloningand potential clinical applications. Thyroid today. 1999;XXII:1-11.
40. Kogai T, Hershman JM, Motomura K, Endo T, Onaya T, Brent GA. Differential regulation of the human sodium/iodide symporter gene promoter in papillary thyroid carcinoma cell lines and normal thyroid cells. Endocrinology. 2001;142:3369-79.
41. Esteller M. CpG island methylation and histone modifications: biology and clinical significance. Ernst Schering Res Found Workshop. 2006;115-26.
42. Peedicayil J. Epigenetic therapy-a new development in pharmacology. Indian J Med Res. 2006;123:17-24.
43. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37-50.
44. Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression on the Na+/I- symporter and iodine accumulation inpoorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metab. 2001;86:3430-5.
45. Riesco-Eizaguirre G, Santisteban P. New insights in thyroid follicular cell biology and impact in thyroid cancer therapy. Endocr Relat Cancer. 2007;14:957-77.
Recibido: 7 de julio de 2010.
Aprobado: 12 de octubre de 2010.
María Teresa Marrero Rodríguez. Instituto Nacional de Endocrinología. Calzada de Zapata, esquina a C, El Vedado, municipio Plaza, Ciudad de La Habana, Cuba. Correo electrónico: mariat.marrero@infomed.sld.cu


{kind=link}