










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol vol.25 no.3 Ciudad de la Habana sep.-dic. 2014
DIAGNÓSTICO
Diagnóstico prenatal de la hiperplasia adrenal congénita, una realidad
Prenatal diagnosis of congenital adrenal hiperplasia
MSc. Tania Mayvel Espinosa Reyes
Instituto Nacional de Endocrinología (INEN). La Habana, Cuba.
RESUMEN
La hiperplasia adrenal congénita es una de las endocrinopatías más frecuentes en la infancia. Resulta desde el punto de vista clínico en un trastorno del desarrollo sexual asociado o no a un cuadro de pérdida salina en la etapa neonatal, manifestaciones de hiperandrogenismo en la adolescencia u oligomenorrea y trastornos de la fertilidad en la adultez. Las posibilidades de diagnóstico en el periodo prenatal han marcado un nuevo hito en el manejo y el pronóstico de estas personas, de ahí el interés por su conocimiento y dominio.
Palabras clave: hiperplasia adrenal congénita, diagnóstico prenatal.
ABSTRACT
Congenital adrenal hyperplasia is one of the most frequent endocrinopathies in childhood. It is from the clinical viewpoint a sexual development disorder associated or not to salt loss condition in the neonatal phase, hyperandrogenism manifestations in adolescence and oligomenorrhea and fertility disorders in the adulthood. The diagnostic possibilities in the prenatal period has marked new milestone in management and prognosis of these patients, hence the interest of professionals for gaining more knowledge about this disorder.
Keywords: congenital adrenal hyperplasia, prenatal diagnosis.
INTRODUCCIÓN
La hiperplasia adrenal congénita (HAC) engloba un grupo de trastornos enzimáticos que conlleva una alteración en la síntesis de cortisol y aldosterona con acúmulo de precursores androgénicos. El déficit de cortisol, que es el hecho común a todas las HAC, por un mecanismo de retroalimentación negativa, produce un aumento de la producción de hormona estimulante de corticotropina (ACTH), y, secundariamente, una hiperestimulación del córtex adrenal, lo cual motiva la elevación de los esteroides previos al bloqueo enzimático. Se heredan de forma autosómica recesiva. Constituyen la causa más frecuente de ambigüedad sexual. Los niños y las niñas son afectados por igual. 1 Las manifestaciones clínicas dependen del déficit enzimático en cuestión, de la severidad del déficit y de la etapa de la vida en la que se presenten, y es el déficit de 21 hidroxilasa (21OHasa). quien es responsable del 90-95 % de las personas con HAC. 1,2
DESARROLLO
Los avances en numerosas especialidades de la Medicina han permitido que, en la actualidad, en un grupo de países desarrollados, se hayan instaurado protocolos para diagnóstico prenatal (DP) de esta enfermedad. Variados son los métodos implementados, la ultrasonografía, la determinación del sexo, y en especial, de regiones específicas del cromosoma Y, estudios bioquímicos en el líquido amniótico, tipificación de antígenos leucocitarios humanos (HLA) e identificación de mutaciones directamente a partir del ácido desoxiribonucleico (ADN) de fragmentos extraídos por punción de vellosidades coriónicas (PVC), todo lo cual ha permitido aumentar la fiabilidad del diagnóstico prenatal.2-7
El primer paso sería definir a quiénes estaría dirigido el DP:
- Gestante con historia personal o familiar de HAC
- Consanguinidad
- Historia de abortos espontáneos
· Antecedentes de muerte neonatal de etiología no precisada
- Asignación de sexo cuestionable al nacer
- Infertilidad
- Síndrome de ovarios poliquísticos (SOP)
Ultrasonografía
La detección por ultrasonido del sexo fetal puede ser de utilidad para decidir si se usan procedimientos invasivos, como la PVC y amniocentesis para cariotipo fetal, cuando se observan alteraciones de la morfología de los genitales externos.
La determinación ultrasonográfica del sexo del feto puede realizarse tan temprano como entre las 12 y las 14 semanas. Según el estudio de Efraty otros,2 en el cual se estudiaron 656 embarazos únicos en los que se realizó ecografía transabdominal en las semanas mencionadas, y se asignó el sexo de acuerdo con el ángulo que formaba el tubérculo genital con una línea horizontal que pasa por la superficie de la piel lumbosacra, visto en un plano medio sagital, se dio la asignación del sexo masculino al feto cuyo ángulo fuera mayor de 30º, y, femenino, cuando el ángulo se encontró paralelo o convergente a la línea horizontal (menos de 10º) (figuras 1 y 2). 8-11
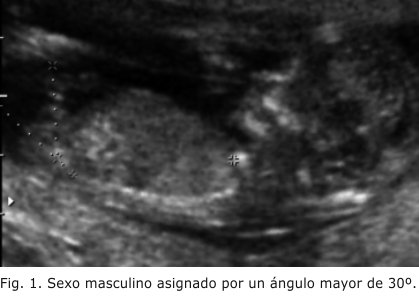
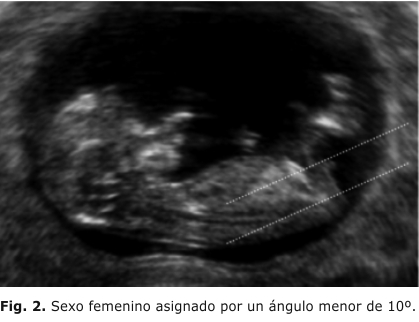
Durante el segundo trimestre, el sexo fetal se puede observar, aproximadamente en el 98 % de los casos. En general, este no será diagnosticado en 10 % de los fetos antes de las 24 semanas, debido a una mala presentación fetal, la posición de la placenta, alteraciones en el cordón umbilical u obesidad materna, entre otras causas.12
Por otra parte, la ecografía de tercer nivel puede sugerir el diagnóstico de genitales ambiguos mediante identificación de vagina y útero en un feto con genitales externos masculinizados.13 Los genitales externos femeninos tienen características ultrasonográficas específicas. Los labios mayores y menores pueden visualizarse desde las 15 semanas de gestación. Un corte axial y coronal de los genitales femeninos demuestra un patrón ecográfico paralelo múltiple, que representa los labios mayores y menores. En la medida que el feto crece, los labios mayores se hacen más prominentes, con una separación central y un eco linear más profundo, que caracteriza los labios.
En relación con la glándula suprarrenal, los hallazgos ecográficos consisten en aumento de su tamaño (diámetro antero-posterior [AP] mayor de 5 mm).13-17
Estudios recientes se refieren a la conservación en la ecogenicidad corteza-médula, con aspecto en patrón cerebriforme, originado en la disposición en pliegues de la glándula redundante, con sensibilidad del 92 % y especificidad del 100 %. Sin embargo, las glándulas suprarrenales de tamaño normal no excluyen el diagnóstico.1,15-18
Determinación del sexo
El diagnóstico del sexo basado mucho tiempo en el cariotipo de células fetales puede realizarse también mediante la amplificación de las regiones específicas del cromosoma Y (gen SRY).La detección de secuencias del cromosoma Y del feto, a partir del ADN extraído del plasma materno, constituye un proceder no invasivo de gran utilidad en aquellas gestaciones con riesgo de HAC por déficit de 21OHasa.
La primera demostración de la presencia de ADN fetal en el plasma materno se hizo utilizando una sonda específica para el gen SRY, localizado en el cromosoma Y.19,20 Desde entonces, la optimización de protocolos basados en la técnica de PCR a tiempo real, ha permitido consolidar esta aproximación como una técnica fiable para la determinación del sexo fetal durante el primer trimestre de gestación.21
En este sentido, la detección del marcador DYS14, presente en forma de múltiples copias en el cromosoma Y, comparado con el marcador clásico SRY (de copia única), ofrece una sensibilidad 10 veces superior, y permite obtener un resultado fiable en semanas de gestación anteriores. 13
El conocimiento del sexo fetal en semanas de gestación tempranas permitiría restringir el tratamiento con corticoides únicamente a gestantes con fetos femeninos, ya que solo en este caso tienen el efecto de evitar la virilización de fetos afectados de esta enfermedad.
Exámenes bioquímicos
- Líquido amniótico : la comprobación de niveles elevados de 17hidroxiprogesterona (17OHP) en el líquido amniótico de la madre con un feto afectado por un déficit de 21OHasa es posible, ya que la producción esteroidea en el feto comienza en la segunda mitad del primer trimestre. La masa suprarrenal aumenta significativamente para esta fecha, y alcanza su punto máximo aproximadamente a las 15 semanas de embarazo. Como la orina fetal contribuye al líquido amniótico desde el segundo trimestre del embarazo, época en la que ya ha comenzado a funcionar la glándula suprarrenal, esto ha permitido identificar fetos con hiperplasia suprarrenal congénita después de la semana 14, lo cual ha abierto el camino del DP de esta afección.18
Otros estudios
- Tipificación de HLA : desde hace algún tiempo quedó establecido que la HAC por déficit de 21OHasa es una enfermedad autosómica recesiva. Se ha demostrado una estrecha relación de la 21OHasa con el locus B del complejo mayor de histocompatibilidad referido al sistema de HLA, por lo que se ha utilizado su tipificación para la identificación de los heterocigotos, y para hacer DP con el análisis de ADN del locus del complejo mayor de histocompatibilidad (HLA) DR o el gen CYP21, por PVC; estos métodos permiten hacer diagnósticos más tempranos que la concentración de la 17OHP en líquido amniótico.18,22
- Caracterización de las mutaciones : la deficiencia de 21OHasa es causada por mutaciones en el gen CYP21 (también llamado CYP21A2). Este se localiza en la región altamente polimórfica del HLA, en el brazo corto del cromosoma 6 (6p21.3), acompañado de un pseudogen CYP21P (CYP21A1P), con el que presenta una homología del 98 %, que se sitúan en tándem después de la porción 3’ terminal de los 2 genes que codifican para el cuarto componente del complemento (C4A y C4B).23-26
Por ser la HAC una enfermedad con herencia autosómica recesiva, el paciente debe tener siempre los 2 alelos afectados (el materno y el paterno). Las anomalías del gen son variables, e incluyen desde mutaciones puntuales (que resultan en la síntesis de una proteína con actividad enzimática variable), hasta deleciones (ruptura y pérdida de la información genética). Esta última alteración implicará obviamente, la ausencia completa de actividad 21OHasa. El fenotipo clínico resultará de la combinación de la anomalía en la expresión de los 2 alelos CYP21A2.26-29
Un nuevo método utilizado es la reacción en cadena de la polimerasa de alelos específicos, con la ventaja de que disminuye el tiempo requerido para el diagnóstico.
El desarrollo de las técnicas de biología molecular realizadas directamente a partir del ADN de fragmentos extraídos por PVC, ha permitido aumentar la fiabilidad del DP. Una vez realizado el estudio familiar, el DP es posible mediante la investigación directa de las mutaciones de la familia en cuestión.
El DP, por la caracterización de las mutaciones, ofrece la oportunidad de, en caso de feto afectado, interrupción del embarazo si la pareja lo decide; o de instaurar un tratamiento intraútero, en caso de fetos con cariotipo 46XX, con el propósito de evitar o atenuar la virilización del feto femenino afectado, evitar la asignación sexual inadecuada al nacer, disminuir la necesidad de cirugías reconstructivas genitales, disminuir los efectos psicológicos de la virilización, así como disminuir el efecto de los andrógenos sobre el cerebro femenino. 30
CONSIDERACIONES FINALES
En la actualidad, con el desarrollo tecnológico, de métodos diagnósticos y las posibilidades de brindar tratamientos desde la etapa prenatal, se considera que el momento óptimo para la determinación del riesgo genético, clarificación del estado de portador y la discusión de la disponibilidad de diagnóstico y tratamiento prenatales, es antes de la concepción, basado fundamentalmente en el diagnóstico molecular de la 21OHasa en un laboratorio de referencia.31
REFERENCIAS BIBLIOGRÁFICAS
1. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke PD, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. September 2010;95(9):4133-60.
2. Efrat Z, Perri T, Ramati E, Tugendreich, D, Meizner I. Fetal gender assignment by first trimester ultrasound. Ultrasound Obstet Gynecol. 2006;27:619-21.
3. Saulo MS, Polanía DF, Osorio P, Pérez J. Diagnóstico prenatal de ambigüedad genital, correlación posnatal y revisión de la literatura. Univ Méd. 2008;49(2):259-76.
4. Costa JM. First-trimester fetal sex determination in maternal serum using real-time PCR. Prenat Diagn. 2001;21:1070-4.
5. Lo YMD, Tein MS, Lau TK, Haines CJ. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet. 1998;62:768-75.
6. Nogués M. Aplicación del diagnóstico de ADN fetal en sangre materna. Progr Diag Trat Prenat. 2006;18(3):135-7.
7. Nimkarn S, New MI. Prenatal diagnosis and treatment of congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Nat Clin Pract Endocrinol Metab. 2007;3:405-13.
8. Betancourt S, Cruz M, Martínez S, Ortega A. Glándula suprarrenal: aspecto ecográfico normal y patologías más frecuentes en el periodo prenatal-neonatal. Revista Colombiana de Radiología. 2004;15(3):1586-90.
9. Hernanz-Schulman M, Brock J, Russell W. Sonographic findings in infants with congenital adrenal hyperplasia. Pediatr Radiol. 2002;32:130-7.
10. Barbat B, Bogyo A, Raux-Demay MC. Screening of CYP21 gene mutations in 129 French patients affected by steroid 21-hydroxylase deficiency. Hum Mutat. 1995;5:126-30.
11. Schulze E, Scharer G, Rogatzki A. Divergence between genotype and phenotype in relatives of patients with the intron 2 mutation of steroid 21-hydroxylase. Endocr Res. 1995;21:359-64.
12. Department of Urology. The New York University School of Medicine. The sonographic appearance of normal and abnormal fetal genitalia. J Urol. 1999;162:530-3.
13. Lee HH, Chao HT, Ng HT, Choo KB. Direct molecular diagnosis of CYP21 mutations in congenital adrenal adrenal hyperplasia. J Med Genet. 1996;33:371-5.
14. Cattani OA, Reyes GM, Azócar PM, Soto MJ, Romeo OE, Valdivia VL, et al. Medición de 17-OH progesterona sanguínea en recién nacidos chilenos: antecedentes para implementar un programa de detección neonatal de hiperplasia suprarrenal congénita. Rev Méd Chile.2000;128(10):1113-8.
15. Tajima T, Fujeda K, Nakayama K, Fujii-Kuriyama Y. Molecular analysis of patient and carrier genes with congenital steroid 21-hydroxylase deficiency by using polymerase chain reaction and single strand conformation polymorphism. J Clin Invest. 1993;92:2182-90.
16. Higashi Y, Hiromasa T, Tanae A. Effects of individual mutations in the P-450(c21) activity and their distribution in the patients genomes of congenital steroid 21-hydroxylase deficiency. J Biochem. 1991;109:638-44.
17. Fardella C, Poggi H, Pineda P. A salt-wasting congenital adrenal hyperplasia: detection of mutations in CYP21B gene in a Chilean population. J Clin Endocrinol Metab. 1998;83:3357-60.
18. Sepúlveda AJ. Hiperplasia adrenal congénita. Rev Chil Obstet Ginecol. 2003;68(1):28-31.
19. Fardella C, Poggi H, Soto J. AMutations in the CYP21B gene in a Chilean population with simple virilizing congenital adrenal hyperplasia. J Endocrinol Invest. 2000;23:412-6.
20. Merino P, Bachega T, Céspedes P, Trejo L, Billerbeck AE, Codner E. Utilidad del estudio molecular de CYP21A2 en el manejo prenatal de hiperplasia suprarrenal congénita: detección de dos nuevas mutaciones en Chile. Rev Méd Chile. 2007;135:1450-5.
21. Zimmermann B. Optimized real-time quantitative PCR measurement of male fetal DNA in maternal plasma. Clin Chem. 2005;51:1598-604.
22. Larrosa CS, Feliu RA, Martín CB, París MN, Escribano SJ. Son útiles los valores analíticos neonatales en el diagnóstico de la forma no clásica de hiperplasia adrenal congénita? An Pediatr (Barc). 2008;68(Supl 2):1-378.
23. Gebara E, Fernández MA, Rojas E, Amina A, López MR. Hiperplasia suprarrenal congénita perdedora de sal en varones durante el período neonatal ¿Es posible adelantarse a la emergencia metabólica? Arch Argent Pediatr. 2009;107(4):369-73.
24. Morel Y, André J, Uring-Lambert B, Hauptmann G, Betuel H, Tossi M, et al. Rearrangements and point mutations of P450c21 genes are distinguished by five restriction endonuclease haplotypes identified by a new probing strategy in 57 families with congenital adrenal hyperplasia. J Clin Invest. 1989;83:527-36.
25. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245-91.
26. White PC, Vitek A, Dupont B, New MI. Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc Nat Acad Sci USA. 1988;85:4436-40.
27. Harada F, Kimura A, Iwanaga K, Shinozawa J, Sasazuki Y. Gene conversion-like events cause steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. Proc Nat Acad Sci USA. 1987;85:8091-4.
28. Higashi Y, Tanae A, Inoue H, Fujii-Kuriyama Y. Evidence for frequent gene conversion in the steroid 21-hydroxylase P-450 (C21) gene: implications for steroid 21-hydroxylase deficiency. Am J Hum Genet. 1988;42:17-25.
29. Wedell A, Thilén A, Ritzén M, Stengler B, Luthman H. Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J Clin Endocrinol Metab. 1994;78:1145-52.
30. González-LM. Mesa Redonda: Actualizaciones pediátricas. Screening metabólico neonatal expandido. Bol Pediatr. 2008;48:329-31.
31. Huidobro Fernández B, Roldán Martína MB, Rodríguez Arnaoa MD, Ezquieta Zubicarayb B. Consejo genético en la hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. An Pediatr. 2012;76(1):51-2.
Recibido: 14 de septiembre de 2013.
Aprobado: 13 de enero de 2014.
Tania Mayvel Espinosa Reyes. Instituto Nacional de Endocrinología. Calle Zapata y D, Vedado, municipio Plaza de la Revolución. La Habana, Cuba. Correo electrónico: tania.espinosa@infomed.sld.cu

