










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol vol.27 no.2 Ciudad de la Habana mayo.-ago. 2016
Rev Cubana Endocrinol. 2016;27(2)
PRESENTACIÓN DE CASO
A propósito de un caso de paraganglioma
A propos of a case of paraganglioma
Valentina Acosta-Ramón,I Maite Estíbaliz López de Goicoecha-Saiz,II Emilio Pariente-RodrigoI
ICentro de Salud “José Barros”. Camargo, Cantabria, España.
IIHospital Sierrallana. Torrelavega, Cantabria, España.
RESUMEN
Los paragangliomas son tumores neuroendocrinos que surgen de los paraganglios autonómicos extraadrenales, los cuales son pequeños órganos formados por células derivadas de la cresta neural embrionaria con capacidad de secretar catecolaminas. Los paragangliomas están estrechamente relacionados con los feocromocitomas porque son indistinguibles a nivel celular, y a menudo comparten las mismas manifestaciones clínicas, como hipertensión, cefalea episódica, sudoración y taquicardia. El diagnóstico de estos tumores es importante por su riesgo de malignización, por las implicaciones de otras neoplasias asociadas, y para la posibilidad de realizar estudios genéticos para detección de otros casos dentro de una misma familia. El objetivo de este artículo es desarrollar un resumen sobre la epidemiología, manifestaciones clínicas, pruebas diagnósticas y tratamiento de estos tumores. Se presenta el caso de un joven de raza negra diagnosticado de un paraganglioma.
Palabras clave: paraganglioma; feocromocitoma; tumores neuroendocrinos.
ABSTRACT
Paragangliomas are neuroendocrine tumors emerging from the extra-adrenal autonomic paraganglia, which are small organs formed by embryonic neural crest-derived cells with catecholamine-secreting capacity. Paragangliomas are closely linked to pheochromocytomas because they cannot be differentiated at cell level and often share the same clinical manifestations such as hypertension, episodic headache, sweating and tachycardia. The diagnosis of these tumors is important because of risk of becoming malignant, the implications of other related neoplasias and the possibility of making genetic studies to detect other cases in the same family. The objective of this article was to make an abstract about epidemiology, clinical manifestations, diagnostic tests and treatment of these tumors. This is the case of a young Black female who was diagnosed with paraganglioma.
Keywords: paraganglioma; pheochromocytoma; neuroendocrine tumors.
INTRODUCCIÓN
Los feocromocitomas y los paragangliomas son tumores neuroendocrinos que pueden producir, almacenar y secretar catecolaminas, sin diferencias entre ambos a nivel celular o de presentación clínica. Las manifestaciones clínicas son consecuencia de un exceso de catecolaminas circulantes, e incluyen: hipertensión (HTA), taquicardia, cefalea, palpitaciones, diaforesis, dolor torácico, ansiedad y pérdida de peso.1 De ellas, la más frecuente es la HTA, que se manifiesta hasta en 90 % de los casos, bien sostenida, o paroxística, en forma de crisis hipertensivas que pueden llegar a ser graves.1,2
La incidencia anual de los tumores secretores de catecolaminas es de 2-8 casos por millón de personas,2 y se corresponden con 0,05 a un 0,1 % de las causas secundarias de HTA en la población general.3
Se suele hacer una distinción entre feocromocitoma y paraganglioma, basada en la localización anatómica; así, se habla de feocromocitomas en referencia a su aparición en la médula suprarrenal; y de paragangliomas o feocromocitomas extraadrenales, en el caso de tumores originados en los ganglios simpáticos, distribuidos a lo largo del eje paravertebral y paraórtico, desde la base del cráneo hasta el suelo pélvico.3 Los tumores secretores de catecolaminas están formados por células cromafines, poliédricas y pleomórficas, y menos del 10 % son malignos. Son signos de malignidad la presencia de muchas células con aneuploidía o tetraploidía, la invasión local de los tejidos vecinos, o la aparición de metástasis a distancia. Los paragangliomas tienen una tasa más elevada de malignidad que los feocromocitomas.4 En los pacientes con enfermedad maligna la supervivencia a los 5 años es de aproximadamente 50 %.5
Tanto los feocromocitomas como los paragangliomas pueden presentarse de forma esporádica (70 %), o asociados a enfermedades hereditarias (30 %). En este segundo caso, los feocromocitomas forman parte del síndrome de la neoplasia endocrina múltiple tipo 2, la enfermedad de von Hippel-Lindau, la neurofibromatosis de von Recklinghausen y los feocromocitomas/paragangliomas hereditarios.2,6 Respecto a los paragangliomas, se ha propuesto una clasificación sobre bases moleculares, en relación con diversas mutaciones de línea germinal en el complejo enzimático succinato-deshidrogenasa.7
CASO CLÍNICO
Se presenta un varón, de 29 años, de raza negra, proveniente de Senegal, que acude a la consulta de Medicina Interna remitido por su médico de cabecera, por haberle observado —en el curso de una ecografía abdominal indicada para valoración de siluetas renales— un nódulo retroperitoneal paraaórtico izquierdo de 29 mm. Sufre de HTA grado 1 mantenida desde hace 5 años, sin que haya llevado nunca tratamiento médico. Refiere sensación de calor y ligera opresión retroesternal, cuando camina durante un tiempo prolongado, que remiten con el reposo. Refiere, igualmente, molestias en zona lumbar y epistaxis frecuentes, así como cefalea leve ocasional. No consume tóxicos. Desconoce antecedentes familiares de HTA.
A la exploración, presenta un índice de masa muscular (IMC) de 24 kg/m2, tensión arterial (TA) 150/85 mmHg, determinada en reposo con esfingomanómetro, y frecuencia cardiaca (FC) de 73 pulsaciones por minuto a nivel de arteria radial, medida en reposo. La auscultación cardiopulmonar se presenta sin alteraciones, no se aprecia temblor, y los pulsos pedios están presentes.
A su llegada a consulta el paciente aportó las pruebas complementarias siguientes:
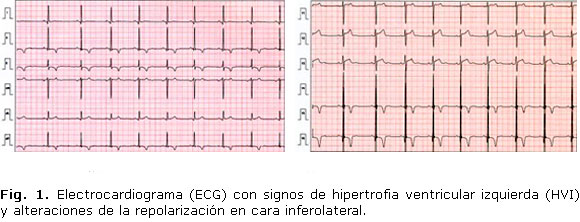
· Electrocardiograma (ECG): ritmo sinusal a 80 latidos por minuto. con signos de hipertrofia ventricular izquierda (HVI) y alteraciones de la repolarización en cara inferolateral (Fig. 1).
· Radiografía de tórax: moderado aumento del índice cardiotorácico a expensas de cavidades izquierdas, sin elongación aórtica.
· Ecocardiograma transtorácico: HVI moderada concéntrica y función sistólica conservada, compatibles con cardiopatía hipertensiva.
· Ecografía renal: siluetas renales sin alteraciones. Nódulo retroperitoneal paraaórtico izquierdo de 29 mm, próximo al hilio renal.
· Analítica sangre: hemograma y bioquímica general básica sin alteraciones. Elemental y sedimento urinario: proteinuria negativa.
El estudio del paciente se completó con los estudios complementarios siguientes:
· Analítica de sangre: Na+ 145 mmol/L, K+ 4,2 mmol/L, Cl- 109 mmol/L, Ca++ 9,9 mg/dL (rango 8,5-10,2), tirotropina (TSH) 2 mU/L (rango 0,4-4,0), tiroxina libre T4L 1,32 ng/dL (rango 0,8-1,9), cortisol basal 19,9 mcg/dL (rango 6-24), aldosterona 117 ng/L (rango 50-200), renina (ARP) 1,3 ng/mL/h (rango 1-2), cociente aldosterona/ARP 9.
· Analítica de orina: noradrenalina 807 mcg/24 h (rango normal 15-80 mcg/24 h), adrenalina 16 mcg/24 h (rango 0,5-20 mcg/24 h), dopamina 755 mcg/24 h (rango normal 65-400 mcg/24 h).
Tras recibir estos últimos resultados se solicitó un estudio de imagen mediante:
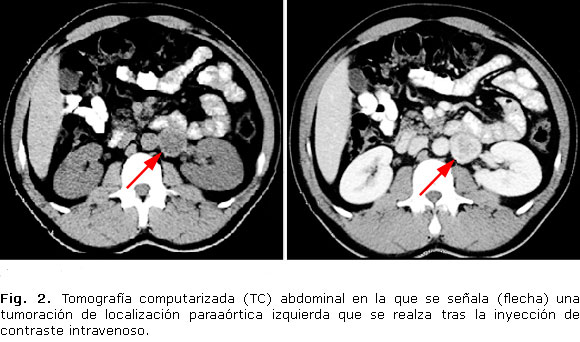
· Tomografía computarizada (TC) abdominal: masa sólida de localización paraaórtica izquierda, situada por debajo del hilio renal, que se realza de forma intensa y periférica con contraste, con diámetros aproximados de 3,7 x 3 x 2,8 cm (longitudinal, transverso y antero-posterior) sugestiva de paraganglioma (Fig. 2).
· Estudio gammagráfico metayodobenzilguanidina marcada con 123I (123I-MIBG) morfofuncionante: zona de hiperfijación a nivel abdominal paramedial izquierdo, compatible con el diagnóstico de feocromocitoma versus paraganglioma. El resto del rastreo corporal fue negativo.
Tras establecer el diagnóstico de sospecha de paraganglioma, se instauró tratamiento con alfabloqueantes (doxazosina), con una dosis de 8 mg repartidas a lo largo de 3 tomas diarias, y se alcanzó un buen control tensional sin precisar betabloqueo. Posteriormente, el paciente fue intervenido mediante laparotomía media infraumbilical, sin que presentara crisis hiperadrenérgicas durante la preparación anestésica ni durante el procedimiento. En el pososperatorio precisó tratamiento con fluidoterapia intravenosa por hipotensión. La biopsia de la pieza resecada confirmó el diagnóstico de paraganglioma (3,5 cm), sin necrosis, mitosis, ni invasión vascular, que no sobrepasa la cápsula tumoral. La evolución posterior del paciente ha sido satisfactoria; se encuentra asintomático, con cifras de TA dentro de la normalidad, sin precisar de tratamiento farmacológico.
DISCUSIÓN
En el paciente la HTA se inició a los 24 años, 5 antes de que se estableciese el diagnóstico de paraganglioma. En este sentido, se ha descrito que, aunque pueden aparecer a cualquier edad, estos tumores son más frecuentes en jóvenes y en adultos de mediana edad.8
Por otro lado, el cuadro clínico asociaba una afectación visceral en forma de cardiopatía hipertensiva, con una moderada hipertrofia concéntrica de ventrículo izquierdo y alteraciones electrocardiográficas en la repolarización de la cara inferolateral. De forma concordante, y según lo publicado, la HTA asociada a un paraganglioma-feocromocitoma, puede ir acompañada de cambios inespecíficos de las ondas ST-T, ondas U pronunciadas, trazados de sobrecarga ventricular izquierda y bloqueo de rama derecha.9
Otras manifestaciones clínicas de nuestro paciente (cefalea ocasional y dolor torácico) son igualmente congruentes. De hecho, además de la HTA, la clínica característica de la hipersecreción de catecolaminas incluye cefalea, palpitaciones, sudoración y palidez. Otros síntomas y signos menos frecuentes son: la astenia, las náuseas, la pérdida de peso, el estreñimiento, la rubefacción y la fiebre. En función de los niveles de catecolaminas circulantes, los pacientes pueden llegar a presentar infarto de miocardio, arritmias, accidentes cerebrovasculares o isquemia en otros órganos.10 El paraganglioma de vejiga urinaria puede manifestarse como crisis hipertensivas típicas al comienzo de la micción y hematuria indolora.
El diagnóstico bioquímico se basa en la determinación de catecolaminas (adrenalina, noradrenadenalina y dopamina), así como sus metabolitos (metanefrina, normetanefrina y ácido vanilmandélico), en plasma o en orina de 24 h. Estas pruebas son diagnósticas en el 95 % de los pacientes sintomáticos,8 si bien hay pacientes con alta sospecha clínica y valores escasamente elevados, en los que se plantea cuál de estas determinaciones es de elección.11 En nuestro caso, los elevados niveles urinarios de noradrenalina y dopamina corroboraron el diagnóstico.
Respecto a la localización del tumor, las técnicas de imagen lo situaron en una zona próxima a la aorta abdominal, por debajo del hilio renal izquierdo. Esta localización es típica, pues la mayoría de los paragangliomas tienen una localización intraabdominal, surgidos del tejido cromafín situado junto a la arteria mesentérica inferior (órgano de Zuckerland), o junto a la bifurcación aórtica.10 Otras localizaciones menos frecuentes son el tórax y la vejiga urinaria. La zona de cabeza-cuello (en general, asociados a los ganglios simpáticos cervicales, o a las ramas extracraneales de los pares craneales IX o X), es la localización habitual de los paragangliomas con menor secreción de catecolaminas, que se presentan como una gran masa asintomática o asociada a hipoacusia unilateral, acúfeno pulsátil o disfonía.12
No se deben iniciar los estudios de localización hasta que las pruebas bioquímicas hayan confirmado el diagnóstico de tumor secretor de catecolaminas.2 Las técnicas de imagen de las glándulas suprarrenales y del abdomen mediante resonancia magnética (RM) o TC, deben ser la primera prueba de localización (sensibilidad superior al 95 %, especificidad mayor del 65 %). Si los resultados de las pruebas de imagen abdominales son normales, está indicada la localización gammagráfica con 123I-MIBG.2 Este radiofármaco se acumula preferentemente en los tumores productores de catecolaminas (sensibilidad del 88 %, especificidad del 84 %), que aparecen como áreas de captación anómala. Aunque se ha sugerido que esta prueba debe realizarse siempre para comprobar la localización y descartar metástasis,13 otros autores consideran superflua la 123I-MIBG en el feocromocitoma-paraganglioma esporádico solitario identificado mediante TC/RM.14
El tratamiento de elección es la resección quirúrgica,3,15 aunque conlleva un alto riesgo de complicaciones perioperatorias, dado que la manipulación del tumor puede dar lugar a la aparición de crisis hipertensivas o arritmias. Estas se previenen mediante el uso de alfabloqueantes, como la fenoxibenzamina o la doxazosina. Una vez establecido el bloqueo alfa, en caso de taquicardia o arritmia, se pueden asociar betabloqueantes como el labetalol.2 Tras la resección tumoral puede aparecer un cuadro de hipotensión arterial severa, que se tratará mediante reposición de volumen. La cirugía del paraganglioma no siempre conduce a la curación del paciente: la HTA persiste en 25 % de los pacientes intervenidos, y se han descrito recidivas hasta en 16 % de los casos, por lo que es preciso un seguimiento clínico y bioquímico indefinido.2,13,16
Se concluye que el caso clínico presentado evidencia el alto índice de sospecha de tumor secretor de catecolaminas en aquellos pacientes con HTA de inicio temprano y hallazgo de una masa abdominal suprarrenal o de localización concordante con paraganglioma. El diagnóstico precoz de estos tumores es relevante por las razones siguientes: 1) la HTA asociada es potencialmente curable mediante la resección quirúrgica del tumor, previniendo el desarrollo de complicaciones vasculares; 2) hasta el 33 % de los paragangliomas tienen un comportamiento maligno,17 y existe la posibilidad de que se asocien a otras neoplasias (síndromes genéticos) que conviene diagnosticar de forma temprana para poder ser tratados; 3) su detección permite llevar a cabo un estudio genético en otros miembros de la familia, para la detección precoz de casos de agregación familiar.
REFERENCIAS BIBLIOGRÁFICAS
1. Pinto A, Barletta JA. Adrenal Tumors in Adults. Surg Pathol Clin. 2015 Dec;8(4):725-49.
2. Young WF. Médula suprarrenal, catecolaminas y feocromocitoma. En: Cecil y Goldman. Tratado de medicina interna. 24ª ed. Madrid: Elsevier; 2013. p. 1474-9.
3. Kiernan C, Solórzano C. Pheochromocytoma and Paraganglioma. Surg Oncol Clin N Am. 2016;25(1):119-38.
4. Mannelli M, Castellano M, Schiavi F, Filetti S, Giacchè M, Mori L, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. 2009;94:1541-7.
5. Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551-6.
6. Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: The expanding genetic differential diagnosis. J Natl Cancer Inst. 2003;95:1196-204.
7. Maher ER, Eng C. The pressure rises: update on the genetics of pheochromocytoma. Hum Mol Genet. 2002;11:2347-54.
8. Bernal C, Alcázar JM. Feocromocitoma: presentación clínica. Diagnóstico y tratamiento. Hipertensión. 2006;23(6):173-83.
9. Landsberg L, Young JB. Feocromocitoma. En: Harrison TR. Principios de Medicina Interna. 14ª ed. Madrid: McGraw-Hill Interamericana; 1998. p. 2337.
10. Herbert C, Sippel R, O'Dorisio M, Vinik A, Lloyd R, Pacak K. The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors: Pheochromocytoma, Paraganglioma, and Medullary Thyroid Cancer. Pancreas. 2010;39(6):775-83.
11. Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287:1427-34.
12. Van Duinen N, Steenvoorden D, Kema IP, Jansen JC, Vriends AH, Bayley JP, et al. Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. J Clin Endocrinol Metab. 2010;95:209-14.
13. Manger WM, Eisenhofer G. Pheocrhomocytoma: diagnosis and management update. Curr Hypert Rep. 2004;6:477-84.
14. Taieb D, Sebag F, Hubbard JG, Mundler O, Henry JF, Conte-Devolx B. Does iodine-131 meta-iodobenzylguanidine (MIBG) scintigraphy have an impact on the management of sporadic and familial pheochromocytoma? Clin Endocrinol. 2004;61:102-8.
15. Cascon A, Pita G, Burnichon N, Landa I, López-Jiménez E, Montero-Conde C, et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J Clin Endocrinol Metab. 2009;94:1701-5.
16. Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med. 2001;134:315-29.
17. Lenders JWM, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366:665-75.
Recibido: 5 de noviembre de 2015.
Aprobado: 3 de enero de 2016.
Valentina Acosta-Ramón. Centro de Salud “José Barros”. Avenida de Bilbao s/n, 39600, Muriedas. Cantabria, España. Correo electrónico: valenacosta84@gmail.com

