










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La diferenciación sexual es un proceso secuencial, en el que se produce una combinación del sexo cromosómico, gonadal, fenotípico y social, cuyo inicio se establece desde el momento de la fecundación1,2,3. El desarrollo sexual adecuado requiere de la presencia de cromosomas sexuales normales (en número y estructura); el desarrollo de las gónadas correspondientes, los conductos sexuales y los genitales externos; y de un medio ambiente hormonal adecuado2. Las distintas alteraciones de estos factores traerán por resultado anomalías en el desarrollo sexual que, en muchos casos, van a traducirse en masculinización incompleta de genitales o grados variables de virilización, de acuerdo con la intensidad y el momento en que se produjo la ruptura de este equilibrio. Se estima que ocurre 1 por cada 5500 nacimientos1.
Cuando el aspecto de los genitales externos es inespecífico y no permite la asignación inequívoca de un determinado sexo, masculino o femenino, se denominan genitales atípicos1. Al referirse a la intersexualidad, los términos usualmente utilizados por investigadores y clínicos incluyen: intersexo, pseudohermafroditismo, hermafroditismo, trastornos de la diferenciación sexual, errores sexuales del cuerpo, defectos de los órganos sexuales, y otros. Sin embargo, existen evidencias de que esta terminología se asocia con sentimientos de vergüenza y aislamiento en pacientes y familiares, por lo que debe evitarse1,2,3. Recientemente se ha propuesto el término “Trastornos del Desarrollo Sexual” (TDS) definidos como estados congénitos en los cuales el desarrollo del sexo cromosómico, gonadal o anatómico es atípico1.
Estas entidades conforman un grupo complejo y heterogéneo. A continuación, se enuncia la clasificación causal de estos trastornos:1,2,3
TDS por alteraciones del sexo cromosómico
45,X (síndrome de Turner y sus variantes).
47,XXY (síndrome Klinefelter y sus variantes),
45,X/46,XY (disgenesia gonadal mixta),
46,XX/46,XY (TDS quimera ovotesticular).
TDS 46,XY (antiguo pseudohermafroditismo masculino)
Desórdenes en el desarrollo gonadal (testicular),
Desórdenes en la síntesis /acción androgénica,
Otras (hipospadias severas, extrofia cloacal).
Presentación del caso
Se presenta el caso de un adolescente masculino de 15 años, con antecedentes de genitales atípicos al nacer, razón por la cual fue remitido al servicio de Endocrinología. A los 10 días de nacido se recibe en la consulta de endocrinología pediátrica del Instituto Nacional de Endocrinología (INEN), en ese momento inscrito como femenino.
Antecedentes patológicos familiares:
Antecedentes prenatales:
Producto de segunda gestación, fisiológica, con seguimiento en consultas prenatales, según programa establecido, que transcurrió con amenaza de aborto no medicada,
Anemia y moniliasis tratadas,
No antecedentes de ingestión y/o exposición a productos hormonales ni tóxicos.
Antecedentes natales:
Parto distócico (cesárea) por presentación anómala,
Peso al nacer (3062 gr),
Talla (50 cm) y circunferencia cefálica (34.5 cm): Normales,
Apgar 9/9,
No asfixia perinatal, no requirió oxigenoterapia.
Antecedentes posnatales:
Caída del ombligo 14 días con granuloma,
Lactancia mixta,
Buen desarrollo psicomotor,
No episodios de vómitos, diarreas o deshidratación.
Al examen físico de recién nacido solo se detectó como alterado la existencia de genitales atípicos: presencia de pliegues labioescrotales, en el derecho se palpaba una tumoración que impresionaba un testículo de 2 ml, y en el izquierdo no se palpaba gónada. Estructura fálica de 1.5 cm con orificio uretral distal. Presencia de orificio vaginal con secreción blanquecina y permeable (sonda que penetra 1.5 cm).
En ese momento se le realizaron complementarios:
Ionograma: Normal,
Medición sérica de 17 OH progesterona: 1 nmol/L (normal),
Ultrasonido abdominal: Órganos abdominales normales, suprarrenales no visibles, no imágenes de genitales internos femeninos,
Ultrasonido cerebral, cardiaco y renal: Normales,
Cistografía miccional: Normal. No presencia de seno urogenital,
Cromatina oral: 0 % de cuerpos de Barr.
Estudio citogenético: En una primera instancia se estudiaron 20 células con resultado de cariotipo 45X, pero se repitió el estudio con 100 células con determinación de sexo cromosómico: mosaico 45X (90 %)/46XY (5 %)/47XYY (5 %), y sexo molecular varón. Cuando aún no se encontraba disponible el resultado de la 17 OH progesterona se inició tratamiento con prednisona 5 mg al día dividido en 3 dosis, ante la posibilidad de hiperplasia adrenal congénita, medicación que posteriormente se suspendió al descartarse esta entidad.
Con los estudios realizados se diagnosticó como un TDS por alteraciones cromosómicas. Se valoró en comisión multidisciplinaria (genética, endocrinología, psicología, urología) y se decidió realizar reasignación de sexo a masculino, con cirugía de reconstrucción genital y gonadectomía izquierda. Se utilizó propionato de testosterona 15 mg intramuscular mensual por 3 dosis, lográndose crecimiento del pene en longitud. El acompañamiento psicológico fue permanente durante esa etapa.
Regresó a consulta con 12 años, preocupado por su desarrollo pondoestatural. Al interrogatorio se comprobó identidad psicosexual masculina, y no se identificó ninguna sintomatología clínica. Según la talla materna (163 cm), y la talla paterna (166 cm), se determinó como talla media esperada (TME) 171cm, la que se ubica entre 50 - 75 percentil.
Examen físico:
Como elementos a señalar en el examen físico se observa la presencia de estigmas turnerianos: ojos grandes almendrados con pestañas largas, nevos pigmentados, implantación baja de pabellones auriculares, y muy discreto pterigion en el cuello.
Genitales externos:
Apariencia masculina,
Teste derecho descendido de 4-5 ml.,
Teste izquierdo no palpable,
Pene con orificio uretral distal, de 2cm.,
Tanner I.
Antropometría
Peso 30 Kg y talla 136 cm. Valoración por las tablas cubanas de crecimiento y desarrollo: Peso/Edad: 10 - 25 percentil (11 años) T/E: 10 percentil (10,2 años). P/T: 50 - 75 percentil (normopeso). Al analizar la curva de crecimiento en el anexo 1 se observa una caída del canal percentilar, por lo cual se considera una baja talla dinámica.
Los complementarios realizados mostraron FSH, LH y testosterona en valores puberales, función tiroidea normal y la exploración de la hormona de crecimiento (GH) también fue normal.
En los estudios de imagen se observa radiografía de silla turca normal. La radiografía de edad ósea se correspondía con 10 años, la cual se diagnosticó como baja talla dinámica y una pubertad incipiente espontánea. Se decidió iniciar tratamiento con GH biosintética (Norditropin 15 click) teniendo en cuenta los siguientes criterios, tener en su fórmula cromosómica un alto porcentaje de líneas celulares correspondientes con 45 X, la disminución del ritmo de crecimiento para esa etapa de la vida y la ubicación en un canal percentilar muy por debajo del esperado según talla media parental (ver señalización en la figura) (Fig.1).
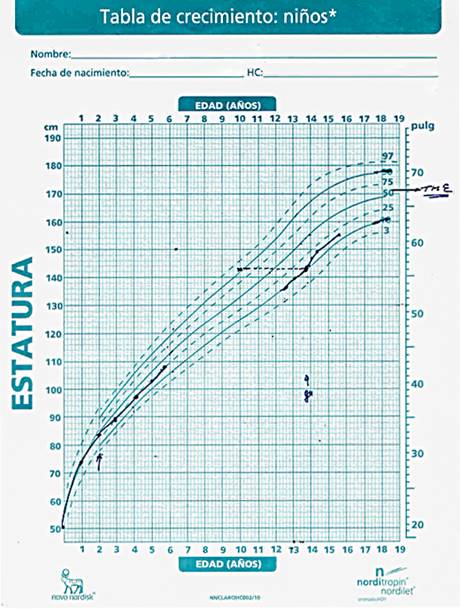
Actualmente el paciente presenta 15 años. La evolución ha sido favorable, con una velocidad de crecimiento normal bajo tratamiento medicamentoso, que logró un ascenso en el canal de crecimiento y alcanzó el estadio final de desarrollo puberal (Tanner V).
Examen físico actual:
Peso 45.5 Kg Talla 155 cm P/E: 25-50 percentil T/E: 10-25 percentil. P/T: 50-75 percentil. (normopeso),
Diámetro biacromial > diámetro bitrocantérico,
Desarrollo muscular de tipo masculino,
Desarrollo puberal: vello sexual y pene Tanner V,
Testículo derecho de 15 ml. Mantiene tratamiento con GH 18 click con interlínea de crecimiento abierta discretamente.
Discusión
La presencia de genitales atípicos al nacer se considera una emergencia social, por la necesidad de identificar al recién nacido y asignar un sexo para la crianza.3 Desde el punto de vista médico, es imprescindible descartar en primera instancia el diagnóstico de hiperplasia adrenal congénita, causa más frecuente de genitales atípicos al nacer.1,2,3 Por el riesgo de presentar una forma clásica de hiperplasia perdedora de sal (potencialmente mortal), con vómitos, diarreas, deshidratación, y desbalance electrolítico debido a la pérdida de sodio que de no ser diagnosticada y oportunamente tratada podría hacer peligrar la vida, se orienta iniciar tratamiento sustitutivo con esteroides hasta obtener la normalidad de los exámenes complementarios.1 El esteroide ideal es la hidrocortisona por ser más fisiológico3, pero cuando se inició el tratamiento del paciente no contábamos con este medicamento, por lo cual se utilizó prednisona.
El mosaicismo implica la coexistencia de dos o más poblaciones celulares de diferentes constituciones cromosómicas, derivadas de un solo cigoto. Para detectar aquellos mosaicismos de baja frecuencia con un intervalo de confianza de un 95 % es necesario el estudio de más de 59 metafases, además de considerar la interfase por FISH (Fluorescence in situ hybridization) para los cromosomas X y Y.4 El primer mosaicismo fue descrito por Ford5 y otros en 1959, quienes observaron que pacientes con síndrome de Klinefelter podían presentar mosaicismo 47,XXY/46,XX. Posteriormente, Fraccaro y otros6por un lado y Ford por otro, observaron casi simultáneamente mosaicismo 45,XO/46,XX en mujeres con síndrome de Turner. A partir de ese momento, otros autores describieron otros tipos de mosaicismo, como XY/XO, XXX/XO, XXX/XX, XXY/XY.7) En 1960 Cooper y otros8describieron un mosaicismo hasta entonces no reportado de tipo XYY/X0, publicado en una revista científica de alto impacto. En 2015 se describe por vez primera9 ) un caso de mosaicismo 45,XO, 46,XY, 47,XYY en una niña de 13 años de edad que consultó por talla baja.
En particular, los mosaicismos por aneuploidías de los cromosomas sexuales son muy infrecuentes. En un análisis de 16 950 casos de diagnóstico prenatal citogenético por amniocentesis, realizados en el laboratorio de citogenética del Centro Nacional de Genética Médica de La Habana durante el período 1984 - 2007, fueron hallados 48 casos de mosaicos cromosómicos, de ellos 18 correspondieron a mosaicos de aneuploidías sexuales,10 ninguno con tres líneas celulares, similar al caso presentado.
En EE.UU. de 600 casos estudiados con aneuploidía del cromosoma Y, solo 24 presentaban el mosaico 45X/46XY/47XYY, 6 casos como diagnóstico prenatal y 18 como posnatal.11
La existencia del cromosoma Y en mosaicismos turnerianos corresponde solo al 2 - 5 % de los casos. En todos los sujetos, la severidad del fenotipo se relacionará con el porcentaje y distribución de cada una de las líneas celulares. Las manifestaciones clínicas resultado de cada fórmula cromosómica en el paciente que se presenta, se exponen a continuación (Fig. 2).
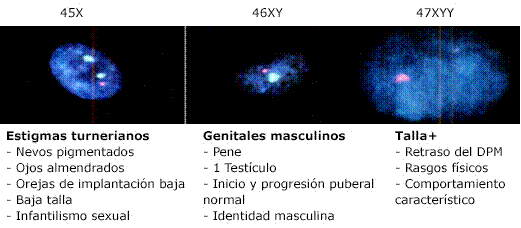
La formación del sexo 45,X/46,XY/47,XYY, puede ser debido al menos por dos mecanismos, el primero es la no disyunción paterna en la meiosis II, seguido por la pérdida del cromosoma en las mitosis subsecuentes. El segundo es un error en la mitosis que explica el diferente número de mosaico en los distintos tejidos.11 El fenotipo de los mosaicismos es muy variable y dependerá del tipo de mutación, el porcentaje de las células afectadas, y la distribución del cambio genético en los tejidos.12 Los casos de mosaicismos de cromosomas sexuales reportados en la literatura muestran gran diversidad.11,12,13,14,15 Los pacientes con sexo cromosómico igual al del caso clínico no siempre se diagnostican en etapa de lactante, debido a que las distintas publicaciones de casos señalan fenotipos que varían desde femenino normal, masculino normal y genitales atípicos, siendo más frecuente el primero. Por ese motivo pasan inadvertidos hasta presentar infertilidad o alguna otra manifestación que requiera de investigaciones complementarias. La manifestación con genitales atípicos es la menos frecuente y se describe su asociación -aunque no en todas las ocasiones- con algunos estigmas turnerianos, debido a la línea celular 45X,11 predominante en nuestro paciente.
La baja talla que se diagnosticó en la adolescencia pensamos es otra manifestación de los rasgos de síndrome de Turner, por lo cual, junto a la posibilidad de crecimiento por interlínea permeable, y la afectación psicológica que pudiera acarrear la baja estatura, se decidió tratamiento con hormona de crecimiento.
El inicio puberal espontáneo y la progresión normal, con identidad psicosexual masculina y valores de testosterona adecuados, se encuentran en relación a la línea 46XY y al rol del sexo molecular del paciente con las implicaciones correspondientes al gen SRY. Aunque, esta línea celular (46XY) solo representaba el 5 %, probablemente, predominaba en las gónadas.
En relación al cariotipo 47XYY en general no traduce un fenotipo típico, aunque se ha mencionado talla ligeramente superior a los hermanos, retraso del desarrollo psicomotor, extremidades largas, acné facial, y comportamiento característico con alteraciones de conducta.3 Ninguno de estos elementos está presente en el paciente.
La inscripción legal de un niño con uno u otro sexo, debe diferirse hasta llegar al diagnóstico definitivo para evitar errores que ocasionen secuelas negativas. En este caso el diagnóstico fue precoz y el tratamiento temprano, el cual, junto con el acompañamiento psicológico y el apoyo familiar, resultó en un adolescente masculino con una buena integración social. La atención óptima de personas con TDS requiere de un equipo multidisciplinario experimentado que debe incluir endocrinólogos pediatras, urólogos/cirujanos, psicólogo/psiquiatra, genetistas, neonatólogos, trabajadores sociales. Otros profesionales de la salud podrían ser requeridos en la atención a recién nacidos con esta condición e incluso sus familiares pudieran necesitar apoyo y acompañamiento durante las diferentes etapas de la vida; en todos los casos es preciso la instrucción, educación y entrenamiento en los aspectos de manejo de los TDS por parte del equipo de salud.
Conclusiones
Los TDS constituyen entidades poco frecuentes en pediatría. Ante la presencia de genitales atípicos al nacer se necesita de un manejo multidisciplinario. El diagnóstico etiológico de los trastornos de la diferenciación sexual es complejo, y requiere de una alta pericia médica. Un tratamiento integral, temprano, con acompañamiento psicológico, y en conjunto con la familia, considerando los principios de la ética médica, les proporciona una buena calidad de vida.

