










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los feocromocitomas son tumores que derivan de las células cromafines adrenomedulares; del 15 al 20 % pueden ser extradrenales, estos reciben el nombre de paragangliomas. La relevancia clínica de ellos se basa en su potencial para secretar catecolaminas, lo que incrementa la morbilidad y la mortalidad cardiovascular.1,2,3,4,5
La mayoría de los feocromocitomas son benignos, pero del 10 % al 15 % pueden ser malignos y, por lo general, estos secretan mayores cantidades de dopamina.6 Estos tumores tienen una elevada incidencia entre los 40 y 50 años de edad, sin que pueda precisarse una diferencia significativa en la frecuencia entre los dos sexos.7 Distinguir si un tumor es benigno o maligno es siempre complejo, pues no existen marcadores histológicos, genéticos, ni moleculares que puedan hacer esta distinción de forma segura.8,9 La malignidad se sospecha en casos con evidencia de invasión local o recurrencia tumoral y/o presencia de metástasis.10,11,12 La malignidad es más común en pacientes con paraganglioma familiar por mutaciones en el gen SDHB,13,14,15 estos pacientes tienen mayor probabilidad de desarrollar neoplasias malignas a otros niveles, como por ejemplo, el carcinoma renal. En cambio, en aquellos con neoplasia endocrina múltiple tipo 2 (MEN2), es más frecuente la etiología benigna.16,17,18,19
La supervivencia en el feocromocitoma maligno a los 5 años, generalmente es del 40 al 70 %.16
En las publicaciones nacionales encontramos cinco reportes de casos de feocromocitoma y uno solo era de etiología maligna.20,21,22,23,24
Esta publicación tiene el objetivo de presentar tres casos clínicos de feocromocitoma maligno, diagnosticados y/o tratados en la sala de hospitalización de adultos del INEN en los últimos 13 años.
Presentación de los casos
Caso 1
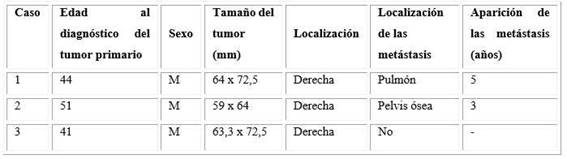
Paciente masculino de 49 años de edad, que ingresa con antecedentes patológicos personales de hipertensión arterial (TA de 12 años de evolución), cumplimiento regular del tratamiento indicado y buen control; y de adrenalectomía derecha, por un feocromocitoma benigno (64 x 72,5 mm) con buena evolución. Cinco años después es hospitalizado por una emergencia hipertensiva −con cifras de TA hasta de 240/120 mmHg− acompañada de cefalea frontal y occipital intensa, palpitaciones y visión borrosa. Además, refirió pérdida de peso y astenia en los últimos meses.
En el examen físico se constató buen control de la TA, con dosis máximas de amlodipino y captopril. En los complementarios se encontró: una diabetes secundaria, territorio de las suprarrenales sin alteraciones, rayos X tórax con múltiples imágenes de + 1 cm, en ambos campos pulmonares; que se confirman en la tomografía computarizada (TC) de pulmón (imágenes sugestivas de metastásis en ambos campos pulmonares). El paciente se remite al servicio de Oncología para su seguimiento y fallece al año del diagnóstico de la enfermedad metastásica.
Caso 2
Paciente masculino de 54 años de edad, antecedentes patológicos personales de diabetes mellitus hace 3 años, con tratamiento regular y buen control y una adrenalectomía derecha hace 3 años, con diagnóstico de feocromocitoma (imagen de 59 x 64 mm). El cuadro se acompañó de hipertensión arterial (HTA) secundaria, con buen control en el posoperatorio sin necesidad de continuar tratamiento antihipertensivo. El paciente es hospitalizado en nuestro servicio porque 2 años después de la cirugía, comenzó con cifras elevadas de TA, acompañadas de palpitaciones y diaforesis, y escalofríos (ante esfuerzos físicos), así como dolor abdominal de moderada a gran intensidad, de localización difusa e irradiado a cadera derecha y región lumbosacra homolateral, con presencia de aumento de volumen de la cadera y limitación para la deambulación. Refirió, además, pérdida de peso de unas 20 libras en 4 meses y constipación.
Al examen físico se detectaron mucosas hipocoloreadas, ligero tinte ictérico, frecuencia cardiaca (FC) de 120 latidos/min, TA de 170/110 mmHg. A nivel del abdomen presentaba dolor a la palpación superficial y profunda a nivel de epigastrio, hipocondrio y flanco derechos; donde se encontró un aumento de volumen de bordes no precisos y con aumento de consistencia y sensibilidad, adherido al tejido circundante. En el sistema osteomioarticular se constató aumento de volumen de la articulación de la cadera derecha, sin signos flogísticos y con dolor a la movilización; y a nivel del sistema nervioso central, se observó disminución de la fuerza muscular en miembros inferiores y alteración de la deambulación por el dolor.
En los complementarios realizados se diagnostica: anemia leve (11 g/dL), eritrosedimentación acelerada (39 mm/h), y glucemia en 8,5 mmol/L. El ultrasonido (US) de cadera informa aumento del líquido a nivel de la articulación de la cadera derecha, compatible con sinovitis, y la TC de pelvis muestra la presencia de una masa de 183 x 113 mm, hiperdensa (35-52 UH) desde la fosa iliaca derecha hasta región glútea con osteólisis de cresta iliaca, porción derecha del sacro y de la articulación coxofemoral derecha, comprimiendo el musculo psoas derecho, con la impresión de enfermedad metastásica. Se ajusta el tratamiento antihipertensivo hasta lograr el control de la tensión arterial y se remite a Oncología para tratamiento paliativo. Fallece a los 6 meses.
Caso 3
Paciente masculino de 44 años de edad, antecedentes de hipertensión arterial, acompañada de cefalea frontooccipital, diaforesis y palpitaciones de difícil manejo. Ingresa en sala de Endocrinología para estudio después de una caída con pérdida de la conciencia, y tensión arterial sistólica hasta de 280 mmHg. En estudio complementario se detecta, en US de abdomen, una imagen tumoral de suprarrenal derecha sospechosa de feocromocitoma.
Clínicamente se mantuvo controlado de la TA, sin otros síntomas positivos con un examen del fondo de ojo con retinopatía hipertensiva grado IV. En los complementarios realizados se encuentran dos glucemias mayores de 7 mmol/L (diabetes probablemente secundaria) y creatinina elevada (174 umol/L). Se realiza TC de abdomen que confirma la presencia de imagen en suprarrenal derecha de 63,3 x 72,5 mm. Se interviene quirúrgicamente y en la biopsia se informa un feocromocitoma maligno. Hasta el momento se mantiene en seguimiento por el servicio de Oncología, con buena evolución clínica e imagenológica. En la tabla se resumen algunas características de los casos.
Discusión
Los feocromocitomas son tumores que, en ocasiones, permanecen sin diagnóstico hasta que los pacientes hacen un cuadro severo de arritmias, infarto del miocardio u otro accidente vascular. Otra forma de presentarse, si no se diagnostica y trata tempranamente, es a través de sus metástasis en los casos de enfermedad maligna. A través de los años, el conocimiento y percepción de estos tumores ha crecido y ha cambiado por los nuevos descubrimientos genéticos, metabolómicos, proteómicos, diagnósticos, terapéuticos y de seguimiento. Recientemente se han descubierto nuevos genes susceptibles con mutaciones somáticas y de líneas germinales y esto pudiera modificar en el futuro las conductas y la evolución de los casos.25
La mayor incidencia del feocromocitoma maligno se encuentra en sujetos entre 40 y 50 años de edad, sin diferencias entre hombres y mujeres..7
En el caso de nuestros pacientes, todos fueron del sexo masculino y de edad media, coincidiendo esta última característica con lo reportado en la literatura.7,24
Estas características de sexo y edad constituyen factores de riesgo importantes de progresión de la enfermedad maligna (sexo masculino y edad mayor de 50 años). Otros elementos que también incrementan la frecuencia de la naturaleza maligna son el tamaño tumoral mayor de 5 cm, la presencia de enfermedad metastásica al momento del diagnóstico, y la localización extraadrenal.26,27,28. Sin embargo, en su estudio Dhir y otros29 encontraron mayor riesgo de malignidad en edades más jóvenes; otros factores como el mayor tamaño tumoral y el diagnóstico de PGL coincidieron con lo reportado por otros autores en su asociación a la malignidad tumoral (p < 0,05). En esta investigación, los pacientes con feocromocitoma metastásico que tuvieron pruebas genéticas negativas y ausencia de historia familiar, tenían tumores iguales o mayores de 4 cm.
En nuestra serie de casos los tumores fueron mayores de 5 cm y localizados en la suprarrenal derecha. En la paciente presentada por Pérez Díaz y otros,24 el tumor también se localizó en la suprarrenal derecha, pero tuvo un mayor tamaño, quizás relacionado con la forma de presentación que se correspondió con un incidentaloma, ya que la paciente acudió por dolor lumbar (10,3 x 6,8 cm).
En cuanto a las diferencias en el sexo de estos tumores, Zelinka30 y otros no encontraron diferencias de frecuencia entre mujeres y hombres; y los pacientes con enfermedad maligna tuvieron una edad media menor y mayor tamaño tumoral cuando se compararon con pacientes con feocromocitoma benigno (41,4 vs. 50,2 años y 8,38 vs. 6,18 cm, respectivamente).
Desde el punto de vista clínico, más del 50 % de los feocromocitomas malignos pueden ser no funcionantes. Los funcionantes pueden tener el mismo patrón clínico que los benignos y manifestarse con la tríada de síntomas clásicos (cefalea, taquicardia y sudoración).31,32,33 Aproximadamente el 60 % de estos casos puede tener hipertensión arterial paroxística34 y en el 82 % esta puede ser más severa,35 como ocurrió en dos de nuestros pacientes.
Distinguir si un tumor es benigno o maligno es siempre complejo, pues no existen marcadores histológicos, genéticos ni moleculares que puedan hacer esta distinción de forma segura.8,9) Por ello, existen diversos sistemas de clasificación con un poder predictivo limitado y además, los biomarcadores no están lo suficientemente validados para el diagnóstico confirmativo de enfermedad metastásica. Debido al pleomorfismo, la atipia nuclear, las figuras abundantes de mitosis e incluso la invasión capsular que puede observarse en feocromocitomas benignos y malignos, el diagnóstico diferencial histopatológico se convierte en un problema para definir el seguimiento y pronóstico del paciente.1,36,37,38).
En los tumores malignos sin evidencia de invasión local, la malignidad se puede sospechar por la presencia de metástasis. En el feocromocitoma los sitios más frecuentes de invasión metastásica son: los ganglios linfáticos (80 %), los huesos (50-70 %), el hígado (50 %), los pulmones (30-50 %) y los riñones.1,10,11,12
Para predecir el riesgo de propagación metastásica se han desarrollado varios sistemas de puntuación sobre la base de la histopatología (actividad mitótica, invasión capsular o vascular, entre otros) y/o de la clínica, pero ninguno de ellos ha demostrado ser confiable, además de las variaciones inter e intraobservador que se describen.39,40
El curso natural del feocromocitoma maligno es aún desconocido. Según la literatura revisada, aproximadamente en la mitad de los casos las metástasis se diagnostican en el momento del diagnóstico; en el resto de los pacientes, estas pueden aparecer unos 5 a 8 años posteriores al tratamiento quirúrgico.10,26,41,42,43Zelinka y otros,30 en 41 sujetos con feocromocitoma maligno, encontraron un tiempo medio de aparición de enfermedad metastásica de 3,6 años y el mayor tiempo reportado fue de 24 años.
Por lo tanto, se sugiere que los pacientes con historia de feocromocitoma, deben ser seguidos, al menos, durante 10 años posteriores a la cirugía. En los casos de alto riesgo (mutaciones germinales, edad menor de 20 años y tumores mayores de 5 cm) se recomienda seguimiento durante toda la vida, con periodicidad anual.44
De preferencia, los pacientes se seguirán con tomografía computarizada contrastada, por su excelente resolución espacial para tórax, abdomen y pelvis. El estudio con resonancia nuclear magnética se reservará para pacientes con enfermedad metastásica, pacientes con paraganglioma craneocervical, con clips quirúrgicos que generen artefactos a la tomografía axial computarizada, con alergia al contraste, y cuando se desee evitar la exposición a la radiación.5
No obstante, aún existen dudas acerca de las características del seguimiento en estos pacientes y se insiste por algunos en el seguimiento personalizado. Basado este en el estudio genético según los datos clínicos, los antecedentes familiares, el fenotipo bioquímico y la localización del tumor. Estudio genético que se recomendaría en el caso de paraganglioma, feocromocitoma adrenal bilateral y unilateral e historia familiar de feocromocitoma/paraganglioma y en todos los casos sugestivos de un síndrome familiar.45
El médico que sigue la evolución de un paciente debe pensar que todos los pacientes con una historia de feocromocitoma o paraganglioma tienen riesgo de recurrencia, a pesar de la resección completa del tumor y que todos los tumores pueden tener un potencial metastásico. Esto es particularmente importante para los tumores mayores de 5 cm. Es más, la localización extraadrenal, el fenotipo bioquímico noradrenérgicoo dopaminérgico, los niveles elevados de cromogranina A, la edad menor de 20 años, la multiplicidad y lo más importante, las mutaciones germinales relacionadas con el grupo 1 pseudohipóxico (especialmente SDHA/B), son asociados con un mayor riesgo de metástasis y un peor pronóstico una vez que estas aparecen.44,45
No obstante se reporta que, aproximadamente en el 50 % de los casos, la enfermedad progresará lentamente (esperanza de vida superior a los 20 años), y en la otra mitad de los pacientes, será de forma rápida, con fallecimiento entre 1 y 3 años, después del diagnóstico.16
En una revisión sistemática, Amar y otros46) estudiaron los factores asociados al riesgo de recurrencia de la enfermedad y encontraron un riesgo menor que lo estimado en estudios previos. Sin embargo, este no fue insignificante. Aproximadamente el 5 % de los casos recurrió en los primeros 5 años de seguimiento; y los últimos eventos se sucedieron hasta 15 años después de la cirugía. Los paragangliomas y la enfermedad familiar fueron los principales factores de riesgo independientes de la recurrencia; y los autores sugirieron la necesidad de incluir la estratificación del riesgo de los pacientes en los protocolos de seguimiento.
Sobre el tema, la guía de la Sociedad Europea de Endocrinología (European Society of Endocrinology, ESE por sus siglas en inglés) publicó los resultados de una base de datos (n = 1153) de seis centros de la red europea para el estudio de tumores adrenales (European Network for the Study of Adrenal Tumours, ENS@T por sus siglas en inglés). Ellos encontraron un riesgo de recurrencia del 10 %, después de 5 años de la resección total del tumor.44
En dos de los casos presentados, la aparición de las metástasis tuvo lugar entre los 3 y los 5 años posteriores a la adrenalectomía; lo que coincide con lo reportado en los estudios mencionados;44,47 pero es importante señalar que el diagnóstico histopatológico inicial no pudo precisar la malignidad de las lesiones, la que se confirmó con la enfermedad metastásica.
En los últimos años se registra una mayor esperanza de vida en pacientes con metástasis. Ejemplo de lo anterior son los reportes de trabajos de Szalat,48Choi49 y otros. Ellos reportaron un caso cada uno, de 51 y 26 años de edad, respectivamente; con metástasis pulmonares y supervivencia de 18 años. Otros autores también han reportado supervivencias de 21 y 30 años50,51 en pacientes tratados con yodo 131-metayodobencilguanidina (I131 MIBG).
El tratamiento de elección es la cirugía siempre que sea posible. Sin embargo, en los casos silentes hay que buscar un balance cuidadoso ente la cirugía y las comorbilidades del paciente. En los casos de alto riesgo para la cirugía, pueden considerarse opciones menos invasivas, no curativas, pero que pueden lograr el control de los pacientes, como la radioterapia y la radiocirugía (gamma-knife, ciberknife).42
En pacientes con enfermedad hereditaria, la cirugía con preservación cortical debe ser siempre considerada, ya que hay, frecuentemente, un alto riesgo de enfermedad bilateral. Este tipo de cirugía más conservadora no está asociada a una disminución de la supervivencia. En el caso de enfermedad metastásica, será recomendada la resección del tumor primario para disminuir en el paciente el riesgo cardiovascular y los síntomas del exceso de catecolaminas o de la invasión tumoral, y así minimizar el área de la radioterapia. Es más, algunos estudios han mostrado que la resección quirúrgica del tumor primario está asociada con una mejor supervivencia, aun en presencia de enfermedad metastásica.
Adicionalmente, la cirugía de las metástasis puede ser considerada en pacientes oligometastásicos o en aquellos en los que se discuta esta decisión terapéutica, aunque existe poca evidencia. En estos casos también pueden emplearse procedimientos mínimamente invasivos como la ablación por radiofrecuencia, la crioablación y la inyección con etanol. El tratamiento con bifosfonatos o denosumab puede ser empleado en casos con metástasis óseas.42,44,45,47,50
La observación con frecuentes seguimientos puede ser un acercamiento inicial óptimo en pacientes con tumores no funcionantes, especialmente en aquellos sin evidencia de crecimiento y/o síntomas o signos de compresión de estructuras adyacentes.
Como limitación de la presentación de casos clínicos podemos señalar las relacionadas con la obtención de los datos de una fuente secundaria, lo que puede contribuir a los sesgos de información y a la ausencia de algunos datos. No se presentan resultados de catecolaminas y metanefrinas por la no disponibilidad.
Conclusiones
El feocromocitoma maligno es infrecuente y se presenta en la edad mediana. Su confirmación diagnóstica clínica e histopatológica ofrece dificultades, sobre todo cuando no aparecen evidencias de invasión tumoral ni lesiones metastásicas.
La enfermedad metastásica debe sospecharse ante la reaparición de los síntomas que motivaron el diagnóstico inicial, lo que puede suceder años después de la adrenalectomía. Esto sugiere la necesidad de seguimiento periódico en estos pacientes durante toda la vida, a pesar del diagnóstico inicial de lesión benigna.

