










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas v.13 n.2 Pinar del Río abr.-jun. 2009
SOCIEDADES CIENTÍFICAS
Cariotipo de alta resolución en sangre periférica en la Neurofibromatosis 1
High Resolution Karyotype in Peripheral Blood in Neurofibromatosis 1
Miladys Orraca Castillo1, Araceli Lantigua Cruz2, Deysi Licourt Otero3.
1 Dra. Especialista de Segundo Grado en Genética Clínica. Asistente. Centro Provincial de Genética Médica Pinar del Río. Pinar del Río.
2 Dra. Doctora en Ciencias Médicas. Especialista de Segundo Grado en Genética Clínica. Profesora Titular. Centro Nacional de Genética Médica. Ciudad de la Habana.
3 Dra. Especialista de Primer Grado en Genética Clínica. Instructora. Centro Provincial de Genética Médica Pinar del Río. Pinar del Rio.
RESUMEN
La Neurofibromatosis tipo 1 (NF1) es uno de los desórdenes autosómicos dominantes más comunes y está causado por defectos en el gen NF1 situado en el cromosoma 17q11.2. Se realizó un estudio descriptivo y transversal en pacientes con NF1 en Pinar del Río durante el año 2005, con el objetivo de identificar en cariotipos de alta resolución defectos cromosómicos y relacionarlos con las características clínicas-radiológicas encontradas, seleccionando 42 pacientes con NF1, asociado a dismorfias, retraso mental, degeneración maligna, neurofibromas plexiformes y tumores del SNC. El 4,7% presentó cariotipo anormal. Las alteraciones estructurales que involucran el gen de la NF1 conllevan a una mayor severidad clínica de la enfermedad.
Palabras claves: NEUROFIBROMATOSIS/ GEN NF1/ CARIOTIPO.
ABSTRACT
The Neurofibromatosis Type-1 (NF1) is one of the most common autosomal dominant disorders and it is provoked by a defect in the NF1 gene located in chromosome 17q11.2. A descriptive, cross-sectional study in patients carrying NF1 in Pinar del Rio was carried out during 2005 aimed at identifying the high resolution karyotypes of chromosomic defects to relate them to the clinical-radiologic features found, to perform it 42 patients carrying NF1 associated with dysmorphia, mental retardation, malignant degeneration, plexiform neurofibromas and tumors in the CNS were chosen. 4, 7% showed abnormal karyotype. The structural changes involving the NF1-gene lead to a greater clinical severity of the disease.
Key words: Neurofibromatosis, gene-NF1, karyotype
INTRODUCCIÓN
Uno de los retos más importantes para la sociedad actual lo constituye la atención a personas que presentan alguna discapacidad enfocada a la prevención, a mejorar su calidad de vida y a la integración social en igualdad de derechos. En este sentido se realizó en Cuba un estudio psicopedagógico, social y clínico genético de las personas con retraso mental y otras discapacidades durante los años 2001-2003.1 Como resultado de este estudio se encontró en la Provincia de Pinar del Río un elevado número de casos con retraso mental a expensas de una enfermedad genética: La neurofibromatosis tipo 1. Se trata de un trastorno del sistema nervioso que se encuentra en cualquier grupo racial que afecta a ambos sexos por igual. Es el tipo más común de neurofibromatosis, se presenta en uno de cada 200 pacientes con retraso mental. Esta enfermedad tiene una prevalencia de 1 por cada 3000 nacidos vivos.2 Es una enfermedad, cuyo diagnóstico es esencialmente clínico. El gen de la neurofibromatosis (gen NF1) tiene una extensión de 300kb aproximadamente, codifica para una proteína llamada neurofibromina que tiene una secuencia de 2818 aminoácidos, esta proteína se expresa abundantemente en órganos como bazo, riñón, timo, testículo, y en el cerebro específicamente en los procesos dendríticos de las neuronas del sistema piramidal, axones de las células de Purkinje del cerebelo, células de Schawn no mielinizadas y oligodendrocitos. 3-5 Asociar la NF1 con desórdenes como los trastornos cognitivos, la epilepsia y el cáncer de piel, y siendo además la NF1 una enfermedad monogénica en la que los cariotipos de rutina resultan normales, entonces se considera valioso la utilización de técnicas citogenéticas de alta resolución, lo que permitirá incrementar el conocimiento científico de esta entidad y será la base para la introducción y aplicación del diagnóstico molecular en Cuba. Lo anterior motivó la realización de este trabajo en la Provincia de Pinar del Río teniendo como objetivos, identificar en cariotipos de alta resolución en sangre periférica los defectos cromosómicos estructurales en el locus génico del cromosoma 17 en pacientes afectados con NF1 y además relacionar los defectos estructurales del cromosoma 17 con las características clínicas-radiológicas.
MATERIAL Y MÉTODOS
Se realizó un estudio descriptivo y transversal desde enero- diciembre del año 2005 en toda la Provincia de Pinar del Río, a partir de 120 familias registradas con el diagnóstico de NF1, de las consultas del Centro Provincial de Genética de Pinar del Río. La muestra estuvo constituida por 42 pacientes con NF1 de cualquier edad, sexo y color de la piel siempre y cuando cumplían con los siguientes criterios:
CRITERIOS DE INCLUSIÓN.
1- Todo paciente con NF1 y que presente, neurofibromas sin retraso mental.
2- Todo paciente con el diagnóstico de NF 1, retraso mental y que el cuadro clínico se acompaña de uno o más de los siguientes hallazgos:
- Dismorfias de la cara, miembros o genitales.
- Degeneración maligna.
- Neurofibromas Plexiformes.
- Tumores del sistema nervioso central.
3- Los pacientes que cumplían con los requisitos anteriores y que dieron el consentimiento para la investigación.
El universo de pacientes se citó hacia el Centro Provincial de Genética de Pinar del Río y además se visitaron en sus municipios para facilitar el trabajo y la selección de la muestra. Se realizaron entrevistas a padres y familiares para completar los datos y obtener la información necesaria, fundamentalmente la relacionada con los antecedentes familiares y la confección del árbol genealógico, fueron sometidos a un examen físico minucioso.
Una vez seleccionados los casos según los criterios de inclusión se les indicó bajo su consentimiento o el de los padres el estudio citogenético. Se realizó el estudio cromosómico de alta resolución, analizaron y clasificaron de más de 10 metafases, con resolución de 850 bandas. Cada indicador investigado se procesó utilizando representación gráfica. Se empleó el Software Estadístico (Stadistic 6.1) y la hoja de cálculo Microsoft Excel.
RESULTADOS
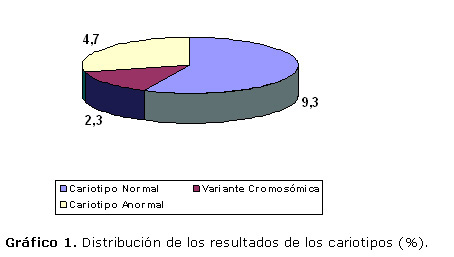
El gráfico 1 muestra la distribución de los resultados cromosómicos, encontrando solo dos pacientes con cariotipo anormal lo cual representa un 4,7%. Se decidió incluir una variante cromosómica normal (inversión pericéntrica del cromosoma 9), que representa un 2,3%.
Correlación genotipo - fenotipo.
- CASO 1
Paciente DRR de 10 años de edad, sexo femenino, raza blanca, producto de un embarazo sin riesgo.
Parto eutócico a las 40 semanas. Peso al nacer: 2940g
Talla: 51cm CC: 35cm CT: 32cm Apgar: 8-9
Edad al diagnóstico: 3 meses Buen Desarrollo Psicomotor
APF: Padre/ sano Madre/ sana Abuelos/sanos
APP: Epilepsia desde un año de edad
Manchas café con leche desde los tres meses de edad.
Síntomas Actuales: Cefalea intensa con mareos y síncopes frecuentes, prurito en todo el cuerpo y disminución de la agudeza visual.
Examen Físico:
1. Manchas café con leche que son incontables y generalizadas.
2. Pecas axilares e inguinales incontables.
3. Desarrollo sexual precoz.
4. Macrocráneo.
5. Fisuras palpebrales antimongoloides.
6. Nódulos de Lisch en ambos ojos en todos los cuadrantes: ojo derecho un número de 14 y ojo izquierdo un número de11.
7. Trastornos del aprendizaje.
Exámenes complementarios:
EEG anormal Rx de cráneo: Displasia del ala mayor del esfenoides.
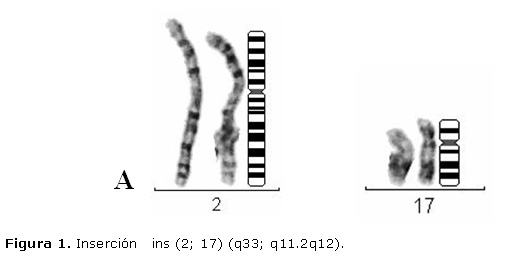
Cariotipo: 46 XX; ins (dois; 17) (q33; q11. 2q12). (Figura. 1).
- Caso 2
Paciente MHM de 40 años, sexo femenino, raza blanca, producto de un embarazo que no refiere conocer detalles de la historia reproductiva, solo que su mamá tuvo 10 embarazos, de ellos los 2 primeros fallecieron sin conocerse el motivo, el resto son normales.
La historia del embarazo no la puede referir.
APF: Hija / NF1 APP: RM Severo, NF1
Edad al diagnóstico: 21 años
Signos al diagnóstico. Manchas café con leche.
Neurofibromas Periféricos.
Síntomas Actuales: Dolor en los neurofibromas, prurito en las lesiones, dolor en ambos miembros inferiores, rodillas y en ambas manos con limitación para los movimientos de flexión y extensión.
Examen Físico:
Hipertelorismo
1. Manchas café con leche que son incontables y generalizadas en todo el cuerpo.
2. Pecas axilares e inguinales incontables.
3. Neurofibromas periféricos incontables y generalizados.
4. Neurofibroma Plexiforme en región lateral derecha del cuello, violáceo de aproximadamente 12 cm de diámetro.
5. Incoordinación motora con trastornos de la marcha.
6. Extremidades: Deformidad de la falange distal del 5to dedo, mano izquierda.
7. RM Severo.
8. Disminución de la agudeza visual (Miopía).
Exámenes Complementarios:
Rx de Cráneo: Signos de displasia del ala mayor del esfenoides. Esclerosis de la pared posterior de las órbitas.
Cariotipo: 46, XX, del 17 (q11.2). (Figura 2).

- CASO 3
La inversión pericéntrica del cromosoma 9, representó el 2,3% de la muestra.
Paciente YPL de 12 años de edad, masculino, raza blanca, producto de un embarazo sin riesgo.
Edad gestacional al parto: 39 semanas.
Parto eutócico.
Parámetros al nacimiento dentro de límites normales.
APF: N/S
APP: NF1
Examen Físico:
- Manchas café con leche incontable y generalizada.
- Pecas inguinales y axilares incontables y generalizadas.
- Escoliosis
- Pectus excavatum.
- Retraso mental leve.
Rx de Cráneo: Disminución de tamaño de ambas órbitas.
Cariotipo: 46, XY, inv (9) (p11; q11)
DISCUSIÓN
Como parte central de la investigación se realizó el estudio citogenético de alta resolución, lo cual ha sido uno de los pasos más importantes de la citogenética humana. Estas técnicas han permitido el descubrimiento de aberraciones cromosómicas sutiles relacionadas con el fenotipo de enfermedades genéticas, oncológicas y otras.
El porciento de cariotipos anormales en este trabajo coincide con otros estudios realizados por Cnossen en 1997 y Kluwe 2004 6,7 que ha sido de un 4,6%. El hecho de que este estudio resulte normal en la mayoría de los pacientes afectados con NF1, no descarta la posibilidad de que presenten una deleción submicroscópica detectable solo mediante técnicas de citogenética molecular.
En el primer caso presentado existe una correlación entre genotipo y fenotipo pero si se tiene en cuenta su edad actual se pudiera pensar que la paciente evolucione hacia la presentación de otros signos más severos de la NF1: como los neurofibromas o tumores del SNC, de forma muy precoz. El rearreglo cromosómico observado en el cariotipo ha sido visto en pacientes con NF1 8-9. Las medidas encaminadas a un asesoramiento genético se imponen y se requiere la confirmación de este hallazgo por métodos de genética molecular.
Con relación al caso 2, los estudios de Venturin y colaboradores 10 establecen una correlación significativa entre pacientes con deleciones 17q11.2 y la presencia de dismorfias faciales entre las que se destaca el hipertelorismo, nariz ancha, fisuras palpebrales con desviación hacia abajo, epicanto, apariencia facial ruda, macrocefalia, además de otros hallazgos como son la baja talla o retardo del crecimiento, el retraso mental, las manifestaciones cardiovasculares y el desarrollo de múltiples neurofibromas. En este caso la paciente presenta algunos signos similares con los descritos en el estudio anterior como son el hipertelorismo, los neurofibromas y el retraso mental.
Se han identificado seis genes candidatos para el retraso mental, en el intervalo de la deleción en pacientes con NF1, de los cuales OMG y CDK5R1 son particularmente interesantes por su función muy conocida en el desarrollo del SNC, los productos de CDK5R1 y el gen p53 forman un complejo presente
en la membrana del complejo de Golgi, por lo tanto su alteración bloquea la formación íntegra de estas membranas, traduciéndose en una desregulación del normal desarrollo neuronal, el gen OMG es un potente inhibidor del crecimiento neuronal, que en conjunto con la desregulación de los factores de crecimiento han sido sugeridos a ser el mecanismo patogénico que explica el retraso mental. 10-14
Se han identificado muchos genes en la región 17q11 que pudieran explicar otras manifestaciones clínicas presentes en esta paciente: 10,15-18
- Gen CCZS (17q11-q12): Produce opacidad congénita de la córnea.
- Gen ERBB2 (17q11.2-q12): Relacionado con el desarrollo oncogénico.
- Gen KRT (17q11-q12): Codifica para la queratina que actúan con genes HOX para el desarrollo dérmico y neuronal.
- Gen SLC6A4 (17q11.1-q12): Transportador de la serotonina.
- Gen VBCH (17q11.2): Relacionado con la hiperostosis cortical generalizada.
Tanto en el caso 1 como en el caso 2 existen criterios clínicos comunes que apoyan la presencia de las aberraciones estructurales encontradas en el cromosoma 17, como son las dismorfias faciales y craneales, los trastornos en el sistema nervioso y en el caso 2 se añaden los neurofibromas.
Si se tiene en cuenta que los pacientes con grandes deleciones frecuentemente sufren las manifestaciones más severas de la enfermedad (anomalías faciales, retraso mental y desarrollo tumoral), que el grupo de pacientes con NF1 con mutaciones intragénicas, entonces se puede tomar en consideración que los estudios de alta resolución en la NF1 pudiera ser indicado a pacientes preseleccionados con esta enfermedad y que presenten además dismorfias faciales, retraso mental, neurofibromas y/o tumores malignos.
La inversión pericéntrica del cromosoma 9, se manifiesta por cambios en la posición del centrómero, constituyendo el 40% del total de las inversiones, por esta razón este rearreglo cromosómico es considerado como una variante normal. Su incidencia oscila entre 1-2% en la población general y habitualmente son heredados, su ocurrencia de novo (como en el caso 3) ha sido reportada en asociación con alguna repercusión fenotípica, que hipotéticamente pueden estar en relación con rearreglos en zonas heterocromáticas con afectación de pequeñas regiones de eucromatina adyacente, la ocurrencia de novo de la inversión pericéntrica del cromosoma 9 ha sido reportada en asociación con retraso mental. 19
En este paciente no se puede, por lo expuesto hasta aquí precisar si existe asociación causal entre ambos fenómenos, lo cual no es raro; pero está presente en este caso la NF1, que cursa con esta complicación.
El diagnóstico temprano de la neurofibromatosis es esencial a fin de que las personas afectadas puedan ser tratadas, asesoradas y referidas a centros especializados, además de facilitar una intervención oportuna en el tratamiento de las complicaciones.
Se puede concluir que el cariotipo de alta resolución en sangre periférica brinda una herramienta más, para el asesoramiento genético en estas familias y finalmente la prevención, ya sea de las complicaciones o de la enfermedad, evidenciado por el porciento de casos positivos encontrados.
Las alteraciones estructurales que involucran el gen de la NF1, conllevan a una mayor severidad clínica de la enfermedad, tal y como se muestra en los pacientes con la aberración cromosómica estructural reportada.
REFERENCIAS BIBLIOGRÁFICAS
1. "Por la Vida": Estudio psicosocial de las personas con discapacidades y estudio psicopedagógico, social y clínico-genético de las personas con retraso mental en Cuba. Ciudad de la Habana: casa editora Abril; 2003.
2. Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrolment. Arch Dermatol. 2005; 141(1):78-9.
3. Gutmann DH, Collins FS. Neurofibromatosis 1. En: Scriver Ch R, Beaudet AL, Sly WS, Valle D dir. The Metabolic-Molecular Bases of Inherited Disease.8th ed. New York: McGraw-Hill; 2001.p. 877-896.
4. Kehrer-Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer V et.al. High frecuency of mosaicism among patients with neurofibromatosis type 1(NF 1) with microdeletions caused by somatic recombination of the JJAZ 1 gene. Am J Hum Genet 2004; 75(3):410-23.
5. Bernheim A, Dessen Philipe, Huret JL, et al.Genes, locations y Genetic Disorders on Cromosoma.Disponible en: http://www.infobiogen.fr/services/chromcancer. 17 monografia en internet acceso el 19 de junio del 2006.
6. Cnossen MH, Vander MN, Breuning MH, Van Asperen CJ, Breslau-Siderius EJ, Halley DJ et al. Deletions spanning the neurofibromatosis type 1 gene: implications for genotype-phenotype correlations in neurofibromatosis type1.Hum Mutat.2004; 9:458-464.
7. Kluwe L, Siebert R, Gesk S, Friedrich RE, Tinschert S, Kehrer-Sawatzki H et al.Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF 1 gene. Hum Mutat.2004; 23(2):111-6.
8. Upadhyaya M, Han S, Consoli C, Majonnie E, H oran M, Thomas NS etal. Caracterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF 1) gene in neurofibromatosis patients with benign and malignant tumors. Hum Mut.2004; 23(2):134-46.
9. Carroll SL, Stonecypher MS. Tumor suppressor mutations and growth factor signalling in the pathogenesis of NF1-associated peripheral nerve sheath tumors: 2. the role of dysregulated growth factor signaling. J Neuropathol Exp Neurol.2005; 64(1):1-9.
10. Venturin MI, Guarnieri PI, Natacci F, Stabile M, Tenconi R, Clementi M et al. Mental Retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Medical Genetics.2004; 41:35-41.
11. Bridge JR, Bridge JA, Neff JR, Naumann S, Ahhof P, Bruch LA. Recurrent chromosomal imbalances and structurally abnormal breakpoint within complex karyotypes of malignant peripheral nerve sheath tumour and malignant triton tumour: a cytogenetic and molecular cytogenetic study. J Clinical Pathology.2004; 57:1172-1178.
12. De Luca A, Bernardini L, Ceccarini C, Sinibaldi L, Novelli A, Giustini S et.al. Fluorescence in situ hibridization analysis of allelic losses involving the longarm of chromosome 17 in NF 1- associated neurofibromas. Cancer Genet Cytogenet.2004; 150(2):168-72.
13. Mueller RF, Young Ian D. Trastornos Monogénicos en Emery´s Genética Médica; 10 ed; Marbán; 2001.p 265-672.
14. Mashour GA, Driever PH, Hartmann M, Drissel SN, Shang T, Scharf B et.al. Circulating growth factor levels are associated with tumorogenesis in neurofibromatosis type 1. Clin Cancer Res.2004; 10(17):5677-83.
15. Huson SM, Rasser EM. The Phacomatoses. En: Emery EH, Rimoins D dir. Principles and Practice of Medical Genetics; 4th ed;United Kingdom: Churchill Livingstone; 2002.Pp.1861-1898.
16. Las neurofibromatosis (monografía en Internet). Nacional Institute of Neurological Disorders and Stroke; Disponible en: http://www.ninds.nih.gov/health and medical/pubs/Las neurofibromatosis.htm. acceso el 22 de julio del 2005.
17. De Luca A, Schirinzi A, Buccino A, Bottillo I, Sinibaldi L, Torrente I etal. Novel and recurrent mutations in the NF 1 gene in Italian patients with neurofibromatosis type 1. Hum Mut.2004; 23(6):629.
18. Lazaro C, Ars E, Serra E, Estivill x. Bases moleculares de la Neurofibromatosis Tipo 1. En: Pascual CI dir. Neurofibromatosis. Madrid: Fundación ONCE; 2001.p.249-251.
19. González GN. Inversión Pericéntrica del 9 y 9qh+. ¿Variantes Cromosómicas? (Tesis para optar por el Título de Master en Genética Médica). Ciudad de la Habana: Centro Nacional de Genética Médica; 2003.
Recibido: 8 de Enero de 2008.
Aprobado: 2 de Junio de 2009.
Dra. Miladys Orraca Castillo. Centro Provincial de Genética Médica Pinar del Río. Pinar del Río.

