










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.15 no.3 Pinar del Río jul.-set. 2011
Feocromocitoma. Presentación de un caso
Pheochromocytoma. A case report
José L. Arteaga Hernández1, Tania Plaza González2, Samael Suero Almonte3, Liván Calzadilla García4, Ceramides Lidia Almora Carbonel5.
1Especialista de Primer Grado en Imagenología. Máster en Longevidad Satisfactoria. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: arteagah@princesa.pri.sld.cu
2Especialista de Primer Grado en Imagenología y Medicina General Integral. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río.Correo electrónico: plaza@princesa.pri.sld.cu
3Especialista de Primer Grado en Medicina General Integral. Residente de tercer año en Imagenología. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: samaelsa@fcm.pri.sld.cu
4Especialista de Primer Grado en Medicina General Integral. Residente de tercer año en Imagenología. Máster en Procederes diagnósticos en APS. Instructor. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río.
5Especialista de Primer Grado Medicina General Integral. Máster en Procederes diagnósticos en APS. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: ceramides71@princesa.pri.sld.cu
RESUMEN
El feocromocitoma es un tumor de la médula adrenal de las células cromafines, derivadas de la cresta neural secretoras de catecolaminas (adrenalina, noradrenalina y menos frecuente dopamina). Se presenta un caso de una mujer joven en el servicio de urgencias del Hospital General Docente Abel Santamaría Cuadrado, con un dolor en el hemiabdomen derecho de varios días de evolución y con cifras de tensión arterial elevada. Se realizó un examen físico exhaustivo, estudios de ecografía abdominal y TAC. Se constató una masa suprarrenal derecha de morfología ovalada y de bordes bien delimitados, que recuerda la imagen de un feocromocitoma; se corroboró por anatomía patológica y confirmó el diagnóstico con características de malignidad. La paciente evolucionó desfavorablemente en el post-operatorio y presentó crisis hipertensivas provocándole la muerte.
DeCS: FEOCROMOCITOMA/ diagnóstico, RADIOLOGÍA.
ABSTRACT
Pheochromocytoma is a tumor of the adrenal medulla in chromaffin cells derived from secretory neural crest of catecholamines (adrenaline, noradrenaline and less frequent dopamine). A case of a young woman attended to the Emergency Room at “Abel Santamaria Cuadrado” hospital is analyzed, the patient complained of a pain in the right hemi-abdomen, of some days of evolution and hypertension. An exhaustive physical examination, abdominal sonography and Axial Computerize Tomography were performed. A right adrenal mass of oval morphology and well-delimited borders, similar to the image of a Pheochromocytoma; pathologic anatomy confirmed the diagnosis with malignant characteristics. The patient showed a favourable post-operative progress but presented a hypertensive crisis which provoked the death.
DeCS: PHEOCHROMOCYTOMA/diagnosis. RADIOLOGY.
INTRODUCCIÓN
El feocromocitoma es un tumor de la médula adrenal de las células cromafines derivadas de la cresta neural secretoras de catecolaminas (adrenalina, noradrenalina y menos frecuente dopamina). Es un tumor poco frecuente (incidencia entre 0,8-2 casos/100.000 habitantes/año), sin diferencias entre sexos y con un pico de incidencia entre la tercera y cuarta década de La alta mortalidad que origina, y el hecho de que la mayoría sean curables quirúrgicamente, hace que sea importante no demorar el diagnóstico. Para su manejo es requisito importante la sospecha clínica y confirmación diagnóstica mediante los exámenes de laboratorio e imagenológicos. En los adultos, el 80% de los feocromocitomas son unilaterales, fundamentalmente derechos, el 10% bilateral, y otros 10% extraadrenales. El 10% de los intraadrenales y el 30% de los de localización extraadrenal son malignos.2
La malignidad viene determinada por la invasión regional y las metástasis a distancia, pudiendo transcurrir largo tiempo desde el inicio de los síntomas hasta el diagnóstico definitivo, presentando la correlación, el tamaño del tumor en el momento del diagnóstico y el tiempo de evolución. El feocromocitoma familiar supone un 5% de los casos, con herencia autosómica dominante y puede aparecer solo o asociado a las neoplasias endocrinas múltiples tipo MEN IIa: S. de Sipple (carcinoma medular de tiroides, hiperparatiroidismo primario y feocromocitomas) o IIb (carcinoma medular de tiroides, feocromocitoma y neuromas fibrosos múltiples), neurofibromatosis de Von Recklinghausen o hemangioblastomatosis tipo von Hippel-Lindau. Los bilaterales se asocian a los síndromes familiares, siendo éstos más metastásicos.3
Como medio de diagnóstico se utilizaron: la radiografía del abdomen, tomografía axial computarizada, que constituye el test de imagen estándar para la localización del tumor, y que puede ser visualizado en la gran mayoría de los casos (>90% de las ocasiones), cuando el tumor supera 1 cm de tamaño la ecografía también se emplea con buenos resultados en la localización del tumor; la resonancia magnética por imágenes tiene una gran rentabilidad en la detección de los tumores, y la ventaja de que permite obtener la imagen longitudinal e identificar mejor los tumores extraadrenales.4 La angiografía puede ser útil cuando el tumor no se localiza con el TAC ni la resonancia magnética por imágenes, aunque nunca es una técnica de primera elección.
Es útil en las masas intraadrenales y extraadrenales que derivan su aporte sanguíneo de la aorta.5 Se propone presentar un caso interesante estudiado en el Hospital General Docente Abel Santamaría Cuadrado.
PRESENTACIÓN DE CASO
Mujer de 33 años, que acude a urgencias por presentar un dolor en el hemiabdomen derecho desde hace varios días. El dolor ha empeorado claramente la noche antes de su visita. Se interpreta como un cólico nefrítico, que no desparece con el tratamiento orientado para con la paciente. Cuando se palpa no presenta peritonismo. No tiene fiebre ni síndrome constitucional. Las cifras de TA son elevadas. La paciente no es diabética y no presenta signos de Cushing ni virilización.
Se realiza ecografía abdominal, TAC: en la TAC se identifican como una tumoración heterogénea con zonas de necrosis y degeneración quística, calcificaciones puntiformes. Figura 1 . masa suprarrenal derecha de 14 x 10 x 10 cm, de morfología ovalada y bordes bien delimitados. Figura 2 : riñón de aspecto normal que además se encuentra desplazado en sentido caudal, anterior y medial.
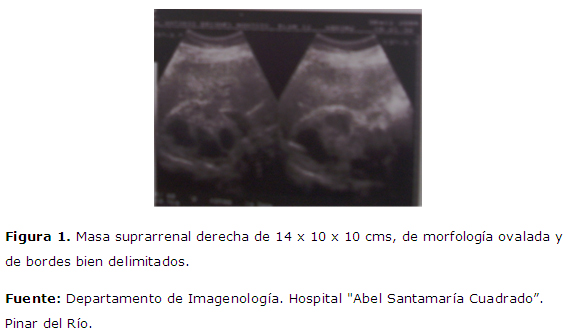
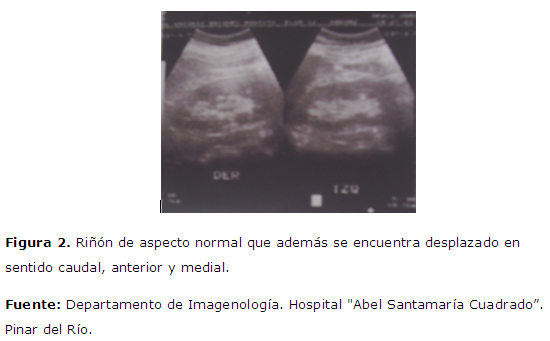
Efectivamente se trata de una masa suprarrenal derecha de 14 x 10 x 10 cm, de morfología ovalada y bordes bien delimitados. El hecho de observar un riñón de aspecto normal, que además se encuentra desplazado en sentido caudal, anterior y medial; la compresión de la superficie lateral de la vena cava inferior y localización retroperitoneal apoyan el origen suprarrenal. Invadiendo estructuras vecinas a la pared posterior de la vena cava, no se observan adenopatías retroperitoneales en otras localizaciones, ni metástasis a distancia. En la TAC se identifican como una tumoración heterogénea con zonas de necrosis y degeneración quística, calcificaciones puntiformes.
DISCUSIÓN
El diagnóstico diferencial por imagen incluye todas aquellas lesiones que con frecuencia comprometen retroperitoneo, dentro de ellos el ganglioneuroma suprarrenal es un tumor casi siempre benigno, formado por las células neurales maduras de los ganglios simpáticos (ganglionares y células de Schwann). Aparece típicamente en los adultos jóvenes. Su lugar más frecuente de localización es el abdomen (retroperitoneo 60% y adrenales 40%). Su aspecto más típico es el de una masa relativamente homogénea, hipocaptante en las fases precoces y que realza, aunque no llamativa en las fases posteriores. Son generalmente bien delimitadas, lobuladas y presentan calcificaciones pequeñas y puntiformes.6
En T1 presentan una señal homogénea e hipointensa, y son hiperintensos y heterogéneos en T2, más hiperintensos a mayor cantidad de tejido mixoide. Los tumores de mayor tamaño pueden contener zonas necróticas y de hemorragia, similares al aspecto del neuroblastoma.7
El neuroblastoma es un tumor de la edad pediátrica, (se origina a partir de células inmaduras de la cresta neural que posteriormente se diferenciaran en células ganglionares del sistema nervioso simpático). El 80% aparecen en menores de 5 años. La edad de la paciente, prácticamente excluye este diagnóstico. Suelen ser masas peor delimitadas y de márgenes no tan precisos, pueden invadir a los órganos adyacentes, metastatizar y producir síndromes paraneoplásicos.8
El feocromocitoma es un tumor de aspecto variado (quístico, con necrosis, calcificaciones y contener grasa) aunque suele ser hipervascular, hiperintenso en T2 e hipointenso en T1.
Estos tres tumores pueden producir catecolaminas y acumular MIBG. La negatividad no excluye el diagnóstico, sin embargo, por las características apreciadas, los hallazgos podrían corresponder con esta variedad de carcinoma suprarrenal; aunque es un tumor muy raro, su máxima incidencia es entre 30 y 70 años, suelen ser grandes en el momento del hallazgo, son además heterogéneos tanto en T1 como en T2 con zonas de necrosis y hemorragia. En este caso, la existencia de invasión de órganos vecinos y de la vena cava inferior hace más probable el diagnóstico.9
En la citopatología, los extendidos son de celulares variables, a veces hemorrágicos. Las células recuerdan a los feocromocitos normales, son poligonales o fusiformes, organizadas en trabéculas y alvéolos relacionados con los vasos sanguíneos, pero otras veces son pleomórficas y multinucleadas. Aunque el criterio inequívoco de malignidad de estos tumores es la metástasis en órganos no cromafínicos, se cita en la literatura algunos hechos que son indicadores de su posible comportamiento maligno tales como: el gran tamaño del tumor; la invasión a tejidos y vasos vecinos; la presencia y número aumentado de mitosis, presencia de necrosis, ausencia de glóbulos hialinos.10
El principal blanco terapéutico es el control de los síntomas secundarios a la excesiva secreción de catecolaminas. El tratamiento actual es quirúrgico con adyuvante de radio fármacos, el cual es rara vez curativo. La mortalidad para los feocromocitomas malignos a los 5 años es mayor al 50 % y no hay un tratamiento específico, por lo que la calidad de vida de los pacientes es el objetivo principal.11, 12 La anatomía patológica confirmó el diagnóstico de feocromocitoma con características de malignidad. La paciente evolucionó desfavorablemente en el post-operatorio y presentó crisis hipertensivas provocándole la muerte.
REFERENCIAS BIBLIOGRÁFICAS
1. Francisco G, Lecube A, Tovar JL, Mesa J. Feocromocitoma extraadrenal maligno: una causa rara de hipertensión arterial. Hipertensión. [Revista en Internet]. 2003 [Citado el 13 de julio de 2011]; 20(2): [Aprox. 2p.]. Disponible en: http://www.elsevier.es/en/node/2023153
2. Stein PP, Black HR. A simplified diagnostic approach to pheocromocytoma. A review of the literature and report of one Institution ´s experience. Medicine. [Revista en Internet]. 1991 [Citado el 18 de julio de 2011]; 70(1): [Aprox. 20p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/1988766
3. Argentina, Academia Nacional de Medicina. Consenso Nacional Inter-Sociedades para el diagnóstico y tratamiento de las neoplasias renales parenquimatosas del adulto. Rev Arg Radiol. [Revista en Internet]. 2010 [Citado el 18 de julio de 2011]; 74(3): [Aprox. 15p.]. Disponible en: http://www.scielo.org.ar/pdf/rar/v74n3/v74n3a11.pdf
4. Ljungberg B, Hanbury DC, Kuczyk MA, Merseburger AS, Mulders PFA, Patard JJ, et al. Guidelines on Renal Cell Carcinoma. European Association of Urology. [Revista en Internet]. 2007 [Citado el 18 de julio de 2011]; 83(2): [Aprox. 11p.]. Disponible en: http://www.uroweb.org/fileadmin/user_upload/Guidelines/08_Renal_Cell_Carcinoma_2007.pdf
5. Renal Cell Carcinoma Staging. Date of origin: 1995. USA: American College of Radiology; 2007.
6. Guía de Recomendaciones para la Correcta Solicitud de Pruebas de Diagnóstico por Imágenes. Argentina: Sociedad Argentina de Radiología; 2008.
7. Sanchez-Recalde A, Costero O, Oliver JM, Iborra C, Ruiz E, Sobrino JA. Pheochromocytoma-related cardiomyopathy (inverted takotsubo contractile pattern). Circulation. [Revista en Internet]. 2006 [Citado el 18 de julio de 2011]; 113: [Aprox. 1p.]. Dispobible en: http://circ.ahajournals.org/content/113/17/e738.full
8. Von Hippel M. Lindau disease germline mutations in Mexican patients with cerebellar hemangioblastoma. J Neurosurg. [Revista en Internet]. 2006 [Citado el 18 de julio de 2011]; 104(3): [Aprox. 5p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/16572651
9. Ramacciato G, Paolo M, Pietromaria A, Paolo B, Francesco D, Sergio P, et al. Ten years of laparoscopic adrenalectomy: lesson learned from 104 procedures. Am Surg. [Revista en Internet]. 2005 [Citado el 18 de julio de 2011]; 71(4): [Aprox. 4p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/15943406
10. Izaki H, Fukumori T, Takahashi M, Taue R, Kishimoto T, Tanimoto S, et al. Indications for laparoscopic adrenalectomy for non-functional adrenal tumor with hypertension: usefulness of adrenocortical scintigraphy. Int J Urol. [Revista en Internet]. 2006 [Citado el 18 de julio de 2011]; 13(6): [Aprox. 4p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/16834641
11. Zacharias M, Haese A, Jurczok A, Stolzenburg JU, Fornara P. Transperitoneal laparoscopic adrenalectomy: outline of the preoperative management, surgical approach, and outcome. Eur Urol. [Revista en Internet]. 2006 [Citado el 18 de julio de 2011]; 49(3): [Aprox. 11p.]. Disponible en: http://www.europeanurology.com/article/S0302-2838%2806%2900029-7/abstract
12. Brukamp K, Goral S, Townsend RR, Silvestry FE, Torigian DA. Rapidly reversible cardiogenic shock as a pheochromocytoma presentation. Am J Med. [Revista en Internet]. 2007 [Citado el 18 de julio de 2011]; 120(9): [Aprox. 1p.]. Disponible en: http://www.europeanurology.com/article/S0302-2838(06)00029-7/pdf/Transperitoneal+Laparoscopic+Adrenalectomy%3A+Outline+of+the+Preoperative+Management%2C+Surgical+Approach%2C+and+Outcome
Recibido:16 de mayo del 2011.
Aprobado: 24 de junio del 2011.
Dr. José L. Arteaga Hernández. Especialista de Primer Grado en Imagenología. Máster en Longevidad Satisfactoria. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: arteagah@princesa.pri.sld.cu.

