










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.16 no.4 Pinar del Río jul.-ago. 2012
Características del neurofibroma plexiforme en pacientes con neurofibromatosis tipo 1. Pinar del Río
Characteristics of plexiform neurofibroma in patients suffering from Type-1 Neurofibromatosis. Pinar del Rio
Miladys Orraca Castillo1, Deysi Licourt Otero2
1Especialista de Segundo Grado en Genética Clínica. Profesora Auxiliar. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico:milgene@princesa.pri.sld.cu
2 Especialista de Primer Grado en Medicina General. Especialista de Primer y Segundo Grado en Genética Clínica. Asistente. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico:deysili@princesa.pri.sld.cu
RESUMEN
El neurofibroma plexiforme es un tumor complejo, que involucra varios tejidos. Llevan a una distorsión masiva del lugar donde se encuentran, originando problemas estéticos y médicos. Con este trabajo se pretende conocer la frecuencia de los neurofibromas plexiformes, en los pacientes con neurofibromatosis tipo 1, la localización, sexo y otras características clínico-genéticas y diseñar un protocolo de diagnóstico y tratamiento de los neurofibromas plexiformes en la provincia. Se realizó un estudio descriptivo, transversal a pacientes, con neurofibroma plexiforme. Del total de pacientes, 34 presentaron neurofibroma plexiforme, predominando el sexo femenino. La mayoría de los neurofibromas plexiformes se localizaron en extremidades, tronco y cráneo-cara. La hiperpigmentación y la hipertricosis resultaron los signos clínicos más frecuentes. Se diseñó un protocolo para el diagnóstico y tratamiento de los afectados.
DeCS: Neurofibroma plexiforme/diagnóstico, genética, complicaciones; Neurofibromatosis/genética, clasificación, diagnóstico.
ABSTRACT
Plexiform neurofibroma is a complex tumor involving several tissues, this tumor provokes a massive distortion of the region where it is located leading to esthetical and medical problems. This work was aimed at knowing the frequency of plexiform neurofibromas in patients suffering from Type-1 neurofibromatosis, location, prevalence of sex and other clinical-genetic characteristics to design a protocol of diagnosis and treatment of plexiform neurofibromas in Pinar del Rio province. A descriptive, cross-sectional study was carried out in patients suffering from plexiform neurofibroma. Out of the total of patients, 34 presented plexiform neurofibroma, female sex prevailed. The majority of plexiform neurofibromas were located in limbs, torso and skull-face. The hyperpigmentation and the hypertrichosis were the most frequent clinical signs. A protocol was designed to accomplish the diagnosis and treatment of the patients affected.
DeCS: Plexiform neurofibroma/diagnosis,genetics,complications; Neurofibromatoses/genetics,classification,diagnosis.
INTRODUCCIÓN
Las neurofibromatosis son un grupo de enfermedades genéticas que se caracterizan por el crecimiento de tumores a lo largo de varios tipos de nervios y que además pueden afectar el desarrollo de otros tejidos tales como huesos y piel. Las neurofibromatosis tipo1 (NF1), o enfermedad de Von Recklinghausen se caracteriza por la presencia de dos o más de los siguientes síntomas: numerosas manchas café con leche, neurofibromas, pecas en axilas o ingles, glioma óptico, nódulos de Lisch y displasia en los huesos largos. La NF1, tiene una expresión muy variable. No se puede predecir la severidad con la que cursará la enfermedad, ni siquiera entre miembros afectados de una misma familia. Actualmente no hay ningún tratamiento curativo, solo existen terapias que pueden mejorar algunos de los síntomas. La NF1 se debe a una mutación genética, es decir, un cambio de alteración en el gen, el cual se encuentra en el cromosoma 17.1-3
La aparición o no de complicaciones no puede predecirse. Dentro de las complicaciones más frecuentes están los problemas de crecimiento, dificultades de aprendizaje, precocidad o retraso de la pubertad, hipertensión, aumento del perímetro cefálico y los tumores.2
El neurofibroma plexiforme es un tumor complejo, usualmente congénito que involucra nervio, músculo, tejido conectivo, elementos vasculares y la piel. Los mismos pueden llevar a una distorsión masiva del lugar donde se encuentran, originando problemas estéticos y médicos graves, de difícil manejo por los especialistas.4
Las lesiones típicas del neurofibroma plexiforme pueden estar presente al nacimiento pero se hace evidente en la mayoría de los casos en las etapas final de la adolescencia cuando el neurofibroma tiene un rápido crecimiento o en la adultez, se han propuesto tres categorías de clasificación de este tipo de neurofibroma de acuerdo a su crecimiento; superficial, displásico e invasivo, estos tumores pueden ser medidos por técnicas de imagen como la resonancia magnética nuclear que también es usada para caracterizar la naturaleza superficial o invasiva del tumor. Ocurren en aproximadamente el 30% de los pacientes con NF1 y de un 7-12% pueden sufrir malignización.4, 5 La ausencia de un protocolo de diagnóstico y tratamiento en Pinar del Río, conllevó a la realización de este trabajo en el cual se describe las características clínicas, genéticas del neurofibroma plexiforme en la provincia.
MATERIAL Y MÉTODO
Se realizó un estudio descriptivo y transversal a los pacientes con NF1 que presentan neurofibroma plexiforme, que asistieron durante el año 2005, a la consulta de Genética Clínica en el Centro Provincial de Genética Médica de Pinar del Río. El universo lo conforman todos los pacientes con NF1 de la Provincia Pinar del Río que constituyen un total de 172, y la muestra todos los pacientes con NF1, que presentaron clínicamente neurofibroma plexiforme.
Todos los pacientes que participaron en el estudio ofrecieron su consentimiento informado para el manejo de la información por parte de los autores encargados de este trabajo, así como para las fotografías que aparecen en el trabajo y la publicación del artículo científico. Se les realizó historia clínica de genética y se incluyeron en el registro automatizado de pacientes con NF1 de la provincia. Se describieron las características clínicas y genéticas fundamentales de los pacientes de la muestra, que incluyeron: la edad de aparición, el sexo, la localización del neurofibroma plexiforme, y los signos acompañantes (dolor, hiperpigmentación, hipertricosis, prurito), casos heredados o de novo, la presencia de otras complicaciones de la enfermedad. Se diseñó un protocolo de diagnóstico y tratamiento de los neurofibromas plexiformes en Pinar del Río. Los resultados se reflejaron en cuadros y gráficos utilizando la hoja de cálculo Microsoft Excel.
RESULTADOS
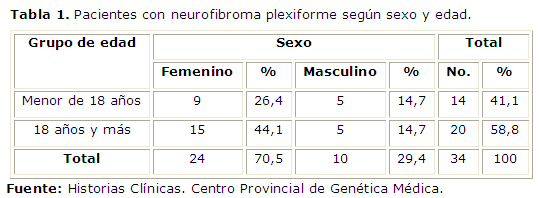
De 34 pacientes diagnosticados con neurofibroma plexiforme, 14 correspondieron a menores de 18 años y 20 de 18 años o más, los cuales se distribuyeron según el sexo, (Tabla 1).
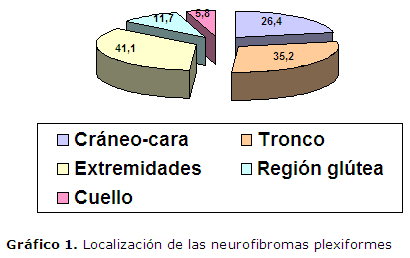
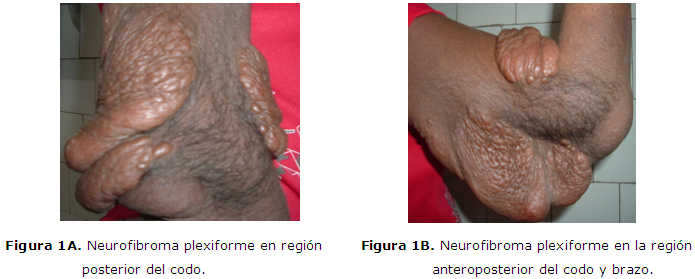
El Gráfico 1 representa la localización de los tumores de este tipo presentes en los 34 pacientes. Predominan los que se encuentran localizados en las extremidades superiores (Figura 1A y 1B) y/o inferiores (41.1%), seguido de los que asientan en la región del tronco ya sea anterior o posterior (35.2%), y le continúan los que se encuentran en el cráneo y la cara, (Figura 2A y 2B), gráfico 2. En algunos pacientes coinciden que los tumores están en más de un sitio.

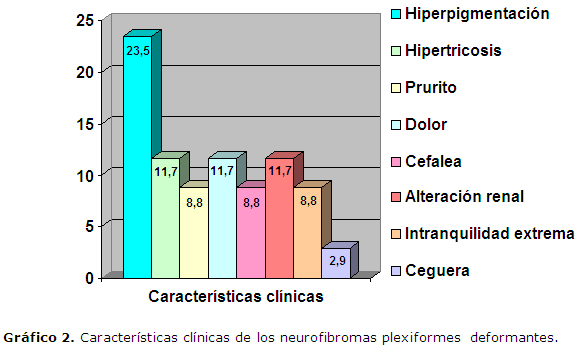
Características como la hiperpigmentación estuvo presente en el 23,5% de los pacientes, la hipertricosis afecta a un 11,7% de los mismos, así como el dolor en el sitio del neurofibroma (Gráfico 2 ).
En este trabajo se estudiaron las características genéticas de los pacientes con neurofibroma plexiforme del total de casos, 19 (55,8%), fueron producto de una nueva mutación y 15 se heredaron de los padres, de ellos 10 se heredaron por vía materna lo que representa un 29,4%, y el resto 14,7% fue transmitido por vía paterna.
La cifoescoliosis, los neurofibromas múltiples y el retraso mental fueron las complicaciones más frecuentes, algunas de estas complicaciones se repiten en un mismo paciente, (Tabla 2 ).

DISCUSIÓN
Los neurofibromas, cuando se extienden por todo el plexo nervioso o parte de él o por varios fascículos de un nervio o por varias ramas de un mismo nervio largo, reciben el nombre de neurofibroma plexiforme, el cual conlleva muchas veces no sólo el engrosamiento de los fascículos nerviosos sino también del tejido de las zonas circundantes, muchas veces con deformidades importantes de éstas.6,7
El mayor número de pacientes con neurofibroma plexiforme pertenece al grupo mayor de 18 años, tabla 1. Generalmente se desarrollan durante la niñez y su crecimiento está limitado. En sus inicios este tipo de neurofibroma puede asemejarse a un tejido suave alargado difuso o a un parche de hiperpigmentación cutánea. Su presencia pasa desapercibida hasta cuando crecen, sobre todo en la etapa de los cambios hormonales, como la pubertad y el embarazo. Es por ello que el tumor deformante se encuentra con más frecuencia en los adultos en la muestra estudiada.6,7 La mayoría de los afectados con neurofibroma plexiforme son del sexo femenino. En bibliografía revisada no se abordan estudios que se refieran a la relación del sexo con la complicación.
Los neurofibromas plexiformes pueden convertirse en masas gigantescas, con formas aberrantes y llegar a comprometer tejidos sanos circundantes. Comúnmente, son tumores congénitos difusos pero a veces son nodulares llegando a desarrollar múltiples tumores discretos. Muchos de ellos crecen internamente, en el tronco y en las extremidades, (Figura 1 A, B, C.)
Pero a veces se presentan alrededor de un ojo, causando un alargamiento en el párpado superior el cual tiende a crecer durante el transcurso de los años. Así mismo, pueden afectar a un lado de la cara. Los afectados con neurofibromas plexiformes en el párpado suelen padecer también de displasia esfenoidal en el mismo lado del ojo afectado. Aunque esta displasia es asintomática, puede asociarse con una hernia a través del defecto óseo.8 (Figura 2 A, B, C)
Las deformidades óseas craneales en la NF1 son progresivas y estos cambios pueden ser explicados por procesos directamente o indirectamente relacionados con la presencia del tumor. Los neurofibromas presentes tempranamente probablemente juegan un rol importante en el desarrollo anormal de las órbitas y el cráneo. Los tumores afectan toda la estructura orbital incluyendo globo ocular, músculos extraoculares, nervio óptico y otras estructuras nerviosas.8 (Figura 2.D)
Se ha publicado la asociación de niveles bajos de vitamina D y la ocurrencia de neurofibromas. El producto proteico neurofibromina es un regulador negativo de las vías de transducción de señales de Ras (Ras forma parte de la cadena de transmisión que va de la membrana plasmática al núcleo en respuesta a factores de crecimiento) por lo que está involucrado en el desarrollo normal del tejido óseo. El tratamiento con vitamina D en estos pacientes inhibe in vitro el crecimiento de líneas celulares. La vitamina D y la neurofibromina pueden interactuar en la reducción de la proliferación celular. La deficiencia de esta vitamina está relacionada con el desarrollo tumoral y su metabolismo interviene en la densidad ósea, osteoporosis, lesiones óseas focales.8
En realidad se plantea que de forma general los neurofibromas plexiformes afectan por orden de frecuencia al tronco, cabeza y extremidades provocando una deformidad estética principalmente en la cara, en este estudio por orden de frecuencia se afecta las extremidades, el tronco y por último la región cráneo-facial, gráfico 1, que esta última localización puede llegar a comprometer el canal auditivo externo y, en consecuencia, afectar la audición, tal y como ocurre con un caso menor de 18 años en este estudio, otro caso mayor de 18 años ha sido operado en múltiples ocasiones por un neurofibroma facial que asienta en el párpado superior. Los plexos preferidos en su localización dentro de la cara, corresponden a los nervios craneales y son las ramas sensitivas del trigémino, las del noveno o neumogástrico y las del décimo o vago, es decir, con afectación preferente de los nervios mixtos, exceptuando el facial que aparece alcanzado raras veces.
El trigémino puede mostrar tumoraciones en todas sus ramas, lo cual lleva a enormes deformidades hemifaciales, con asimetría muy notoria. Sin embargo, el trastorno alcanza especialmente a los nervios de la primera rama trigeminal u oftálmica, con presencia de abultamientos tumorales en la propia órbita, en párpado superior y, muy frecuentemente en región de las sienes y de la frente. La ocupación de la órbita por estos neurofibromas provoca exoftalmos más o menos acusado.9, 10
Como premisa fundamental los casos que se exponen en este trabajo tienen deformación importante de la estructura donde se encuentran los neurofibromas plexiformes. El 19.7% de las pacientes atendidos en el Centro Provincial de Genética Médica de Pinar del Río con NF1 tuvieron neurofibromas plexiformes. Existen reportes que plantean que este tipo de lesiones se observan en el 10-50% de los individuos afectados.
Un neurofibroma plexiforme puede venir acompañado de una hiperpigmentación y/o de hipertricosis, erosión ósea que produce dolor y otros síntomas acompañantes, lo anterior coincide con lo descrito en este trabajo en el que en todos los pacientes se presentan algunos síntomas y signos acompañantes Gráfico 2.5-9 Se reporta en la literatura que los cambios de textura, el dolor persistente, incremento en el tamaño del neurofibroma constituyen hallazgos clínicos de malignidad, por lo que el seguimiento clínico es mandatorio.
La NF1 es una enfermedad que tiene herencia autosómica dominante con penetrancia completa, aunque el 50% de los casos representa una nueva mutación, lo cual coincide con este estudio que reporta un 55.8% de casos producto de una nueva mutación. Las evidencias basadas en análisis de ligamiento, han indicado que la vasta mayoría de las nuevas mutaciones provienen de alelos paternos, indicando que estas mutaciones ocurren aparentemente durante la espermatogénesis. Se reporta en la literatura que los pacientes nacidos de madres afectadas expresan una forma más severa de la enfermedad con relación a los que procedían de padres afectados. A este fenómeno se le denominó impronta genómica, este es el fenómeno por el cual un gen o región de un cromosoma muestra una expresión diferente dependiendo del origen paterno o materno.11 En este estudio de los 15 casos descritos, con neurofibroma plexiforme, que presentaron una forma heredada de la mutación, la mayoría son de origen materno, lo cual coincide con un estudio publicado en esta provincia que describe un 26,1%.
El ciento por ciento presentó al menos otra complicación predominando el retraso mental o trastorno del aprendizaje. La dificultad de aprendizaje aparece aproximadamente en un tercio de las personas con NF1, específicamente en otras palabras pueden experimentar problemas al aprender a leer o escribir o a la aritmética. Es importante para los padres y maestros de estos niños estar atentos. Como los niños no tienen otros problemas en otras áreas como la expresión verbal, pueden ser acusados de ser holgazanes. Pero, sin embargo si los maestros saben que un niño tiene NF1 y que puede tener este problema, pueden ayudar en la necesidad de apoyo específico de estos niños para superarlo. Sumado a los problemas de aprendizaje, se puede notar que estos niños son más "toscos". Ambas cosas, la "tosquedad" y los problemas de aprendizaje tienden a disminuir con la edad y el apoyo apropiado.7, 8
En la literatura se afirma que los tumores plexiformes malignos se originan en tumores neurofibromatosos plexiformes pre existentes por lo que se insiste en la práctica biopsia de un fragmento tumoral aspecto este no logrado en la mayoría de los casos de este trabajo. A pesar de que el tratamiento fundamental de las NF plexiforme sigue siendo quirúrgico existe un gran dilema reflejado en la literatura sobre este aspecto, dado por las complicaciones ocurridas durante el acto quirúrgico, no obstante se ha clasificado la NF atendiendo a tres grupos según el grado de afectación (I afectación exclusiva de partes blandas, II afectación ósea con visión conservada y III afectación ósea con pérdida de la visión) para los cuales se ha prescrito diferentes esquemas de tratamiento quirúrgico.10,11
No obstante, teniendo en cuenta la evolución y características de los casos de este estudio se consideró que se debía diseñar un protocolo útil para el diagnóstico y tratamiento de los casos con neurofibromas plexiformes.
Los neurofibromas plexiformes observados en la muestra estudiada tuvieron una frecuencia diferente, acorde a la edad de los pacientes. Los neurofibromas plexiformes más frecuentes estuvieron circunscritos a las extremidades y el tronco. La mayoría de los que presentaron una forma heredada de la mutación, son de origen materno, estos elementos contribuyen a la calidad con la que se debe brindar el asesoramiento genético de los pacientes y las familias. Se debe dar continuidad a la vigilancia estricta de estos casos, por lo que fue necesario diseñar un protocolo de trabajo para el diagnóstico, manejo y tratamiento de las personas afectadas.
REFERENCIAS BIBLIOGRÁFICAS
1. Joseph H. Hersh, MD. Health Supervision for Children with Neurofibromatosis. Pediatrics [Internet]. Mar 2008 [citado 8 Jun 2012];121(3):[aprox. 12 p.]. Disponible en: http://neoreviews.aappublications.org/content/pediatrics/121/3/633.full.pdf+html
2. Ferner RE. Neurofibromatosis 1. Eur J Hum Genet [Internet]. 2007 [citado 8 Jun 2012]; 15(2):[aprox. 8 p.]. Disponible en: http://www.nature.com/ejhg/journal/v15/n2/pdf/5201676a.pdf
3. Casanova RT, González BC. Neurofibromatosis tipo 1. Medicentro 2008; 12(3): 1-5. http://www.medicentro.sld.cu/paginas%20de%20acceso/Sumario/ano%202008/v12n3a08.htm
4. Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis Type 1 Revisited. Pediatrics [Internet]. Ene 2009 [citado 8 Jun 2012]; 123(1): [aprox. 12 p.]. Disponible en: http://pediatrics.aappublications.org/content/123/1/124.full.pdf+html
5. Dombi E, Solomon J, Gillespie AJ, Fox E, Balis FM, Patronas N, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology [Internet]. Feb 2007 [citado 8 Jun 2012]; 68(9):[aprox. 5 p.]. Disponible en: http://www.neurology.org/content/68/9/643.short
6. Fisher MJ, Basu S, Dombi E, Yu JQ, Widemann BC, Pollock AN, et al. The role of [18F]-fluorodeoxyglucose positron emission tomography in predicting plexiform neurofibroma progression. J Neurooncol [Internet]. 2008 [citado 8 Jun 2012];87(2):[aprox. 7 p.]. Disponible en: http://dx.doi.org/10.1007/s11060-007-9501-5
7. Ferner RE, Golding JF, Smith M, Calonje E, Jan W, Sanjayanathan V, et al. [18F]2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) as a diagnostic tool for neurofibromatosis 1 (NF1) associated malignant peripheral nerve sheath tumours (MPNSTs): a long-term clinical study. Ann Oncol [Internet]. Feb 2008 [citado 8 Jun 2012];19(2):[aprox. 5 p.]. Disponible en: http://annonc.oxfordjournals.org/content/19/2/390.full.pdf+html
8. Jacquemin C, Bosley TM, Svedberg H. Orbit Deformities in Craniofacial Neurofibromatosis Type1. AJNR Am J Neuroradiol [Internet]. Sep 2003 [citado 8 Jun 2012];24(8):[aprox. 5 p.]. Disponible en: http://www.ajnr.org/content/24/8/1678.full.pdf+html
9. Lammert M, Friedman JM, Roth HJ, Friedrich RE, Kluwe L, Atkins D, et al. Vitamin D deficiency associated with number of neurofibromas in neurofibromatosis 1. J Med Genet [Internet]. Sep 2003 [citado 8 Jun 2012]; 43(10): [aprox. 5 p.]. Disponible en: http://jmg.highwire.org/content/43/10/810.full.pdf+html
10. Gachiani J, Kim D, Nelson A, Kline D. Surgical management of malignant peripheral nerve sheath tumors. Neurosurg Focus. Jun 2007; 22(6):13. PubMed PMID: 17613204.
11. Ferner RE. Neurofibromatosis 1. European Journal of Human Genetics [Internet]. 2007 [citado 8 Jun 2012]; 15(2):[aprox. 8 p.]. Disponible en: http://www.nature.com/ejhg/journal/v15/n2/pdf/5201676a.pdf
Recibido: 29 de noviembre de 2011.
Aprobado: 16 de julio de 2012.

{kind=link}