










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.16 no.5 Pinar del Río sep.-oct. 2012
PRESENTACIÓN DE CASO
Epidermolisis bulosa simple. Presentación de un caso
Simple epidermolysis bullosa. A case report
Omar León Vara Cuesta1, Yanett Sarmiento Portal2, María Elena Portal Miranda3, Sergio Piloña Ruiz4, Jesús Juan Rodríguez5 .
1Especialista de Primer Grado en Medicina General Integral y de Segundo Grado en Neonatología. Máster en Atención Integral al Niño. Profesor Auxiliar. Hospital General Docente. "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: belkys@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral y de Segundo Grado en Neonatología. Máster en Atención Integral al Niño. Asistente. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: yanettsp@pricesa.pri.sld.cu
3Especialista de Segundo Grado en Neonatología. Máster en Atención Integral al Niño. Asistente. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: portal23@pricesa.pri.sld.cu
4Especialista de Segundo Grado en Neonatología. Máster en Atención Integral al Niño. Instructora. Hospital General Docente "Abel Santamaría Cuadrado". Pinar del Río. Correo electrónico: sheila@pricesa.pri.sld.cu
5Especialista de Segundo Grado en Pediatría y Genética Clínica. Profesor Auxiliar. Hospital Pediátrico Pepe Portilla. Pinar del Río. Correo electrónico: hildeliza@pricesa.pri.sld.cu
RESUMEN
La epidermolisis bulosa se refiere a un grupo heterogéneo de enfermedades hereditarias ampulosas crónicas, que tienen patrones de herencia y manifestaciones clínicas diferentes, que afectan a la piel y las mucosas con formación de ampollas y vesículas tras mínimos traumatismos, con afectación variable de otros órganos. No existe un tratamiento específico, su evolución es crónica, llegando a mermar la calidad de vida de los pacientes y su supervivencia, lo cual supone un reto tanto para los afectados, como para familiares y profesionales que los atienden. Se presenta el caso de un recién nacido con diagnóstico de epidermolisis bulosa simple de manos y pies y se ofrece una revisión actualizada sobre el tema.
DeCS: Epidermólisis ampollosa/complicaciones.
ABSTRACT
Epidermolysis bullosa is a heterogenic group of chronic-bullosa-inherited diseases having different clinical patterns and manifestations of inheritance that affect the skin and the mucosa with formation of bullae and vesicles after minimal traumas affecting other organs variably. Non-specific treatment is known, its natural history is chronic, observing a reduction of the quality of live in patients and a decline in survival rates; which implies a challenge to patients, families and medical professionals. A case of a newborn baby having simple epidermolysis bullosa in hands and feet is presented, an update review about the topic is proposed.
DeCS: Epidermolysis bullosa/complications.
INTRODUCCIÓN
Las epidermolisis bulosas (EB) congénitas son un grupo complejo de enfermedades genéticas degenerativas de la piel, caracterizadas por no generar la proteína que permite que la dermis se una a la epidermis, lo que produce ampollas con el mínimo roce o sin roce aparente. Tienen la piel tan frágil como "alas de las mariposas", por lo que se les llama "niños mariposa". Las lesiones típicas son semejantes a quemaduras, con la diferencia que en la EB continuamente aparecen lesiones nuevas. Las grandes extensiones de piel afectada aumentan el riesgo de tener infecciones, deshidratación, desequilibrio electrolítico y desnutrición.1-2
Se estima que afecta uno de cada 17.000 nacidos vivos y que en el mundo existen alrededor de 500.000 casos de epidermolisis bulosa, 3 siendo la prevalencia del orden de 32 casos /millón de habitantes, con una incidencia de casos nuevos de 1,4 /de habitantes /año con variaciones que dependen más de la calidad de los registros que de diferencias regionales o étnicas. Se manifiesta por igual en todos los grupos raciales y étnicos, así como en ambos sexos. No es una enfermedad infecto-contagiosa, sino genética y las formas de herencia, pueden ser autosómicas recesivas y autosómicas dominantes.3-4
En la actualidad el diagnóstico específico se hace por el estudio histopatológico con microscopía electrónica y/o convencional, análisis histoquímico y estudio molecular de los genes de las proteínas involucradas.5-6
La epidermolisis ampollosa no tiene curación, aunque algunos subtipos mejoran con el tiempo. El tratamiento es, por lo tanto, paliativo y tiene por objeto aliviar los síntomas cutáneos y extracutáneos de la enfermedad. En todos los casos, es necesario que el paciente mantenga una buena higiene corporal y bucal.7
Se presenta un recién nacido con diagnóstico de (EB) simple atendido en el Servicio de Neonatología del Hospital General Docente "Abel Santamaría" en Pinar del Río y se ofrece una revisión sobre el tema.
PRESENTACIÓN DEL CASO
Recién nacido sexo masculino, de raza blanca, hijo de madre de 24 años de edad, con antecedentes de salud e historia personal y familiar de defectos congénitos, embarazo de bajo riesgo obstétrico, G1, P0, A0, serología no reactiva, grupo y factor A+, producto de parto eutócico a las 36,4 semanas, líquido amniótico claro, cordón y placenta normal, tiempo de rotura de membranas 8 horas, Apgar 9-9 puntos y peso al nacer 3400 gramos.
Al nacer en el examen físico se constató la caída de piel en colgajos a nivel de la nariz, antebrazo izquierdo, mano y rodilla derecha, sospechándose el diagnóstico de epidermolisis bulosa, trasladándose de inmediato al Servicio de Cuidados Especiales Neonatales. (Figura 1).
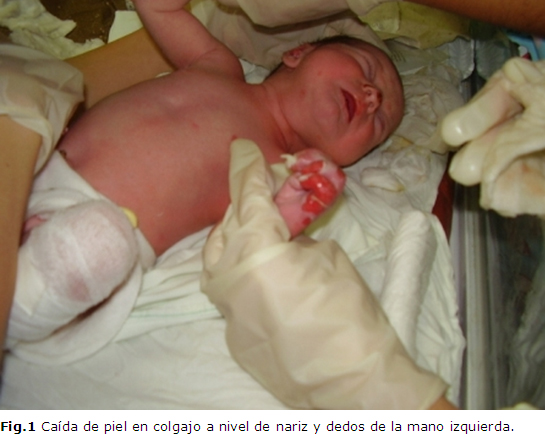
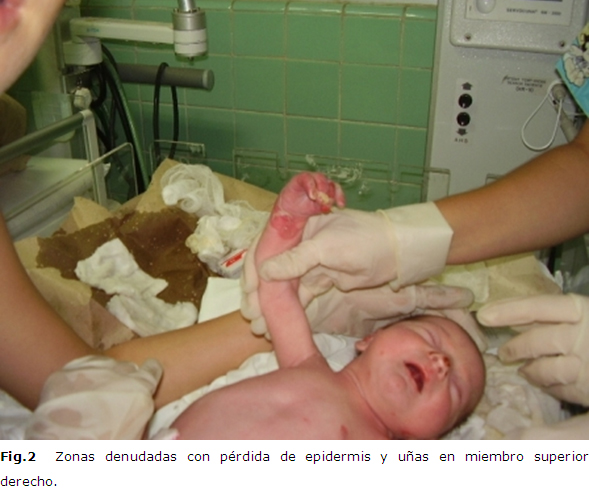
A las 34 horas, se apreció la aparición de nuevas lesiones ampollosas con líquido seroso en su interior a nivel de los dedos de la mano izquierda y glúteos; sucesivamente fueron apareciendo nuevas lesiones en las zonas sometidas a estímulos mecánicos mínimos; al quinto día a nivel del pie derecho se detectó una amplia zona denudada, con perdida de la epidermis y las uñas, con exudación serosa, pero sin signos de infección local en ninguna de las lesiones. (Figura 2)
A pocas horas posteriores a su nacimiento se realizó interconsulta con Dermatología y Genética Clínica planteándose una forma probablemente autosómica dominante (mutación de novo) de epidermólisis bulosa simple de manos y pies, basado en que los padres son fenotípicamente normales y no hay antecedentes familiares de la enfermedad. Tampoco existe consanguinidad y se trataba del primer hijo de ambos sin historia de abortos.
El manejo estuvo encaminado a evitar aparición de nuevas lesiones, logrando manipulación mínima y por personal especializado, así como evitar empeoramiento de las lesiones ya existentes aplicando tratamiento antibiótico tópico con gentamicina 0.1% (crema) y vendajes oclusivos húmedos y suaves, así como fomentos de permanganato de potasio(Mno4K) 1:4000 en las áreas denudadas durante 30 minutos cada 8 horas. Posteriormente se cambio el antibiótico tópico por Mupirocina 2% y se agrego hidrocortisona (crema) al tratamiento.
La evolución fue satisfactoria sin signos de sepsis, y el cultivo bacteriológico de las lesiones fue negativo. Durante la hospitalización se ofreció el asesoramiento genético a los padres y familiares, y se entrenó a la madre en lo relacionado a las curas y manipulación del niño, así como se explicaron detalladamente los cuidados a tener en el hogar en la prevención de complicaciones futuras.
Se egreso a los 26 días con un seguimiento en la consulta multidisciplinaria (Neonatología, Pediatría y Dermatología). Actualmente tiene 6 meses de edad y su evolución clínica ha sido satisfactoria. El riesgo de recurrencia estimado en la pareja como forma autosómica recesiva es de un 25%, pero en los casos de mutaciones «de novo» autosómica dominante el riesgo de recurrencia en la pareja es bajo.
DISCUSIÓN
Existen 20 subtipos de EB que se agrupan en 3 grupos principales dependiendo del lugar de localización de la ampolla. Esta clasificación se lleva a cabo en base a la Clasificación del Registro Nacional de Epidermolisis Ampollosas de 1991, (modificada por Fine en el año 2000), esta clasificación se basa en criterios clínicos, el mecanismo de transmisión, la proteína y gen alterado y su localización cromosómica.1, 3
• EB Simple: la ampolla se localiza a nivel intraepidérmico, justo en las células de la capa basal; cicatrizan sin pérdidas de tejido, mejora con el tiempo. En su mayoría son de herencia dominante y genéticamente está alterada la codificación de las moléculas de queratina formadoras del citoesqueleto 5 y 14, por lo que se producen anomalías estructurales y bioquímicas en la formación de los tonofilamentos que conducen a la desestructuración de las células de la capa basal de la epidermis.3,7
• EB Juntural o de la unión: Las ampollas aparecen entre la epidermis y la dermis, a nivel de la membrana basal en general se deben a mutaciones que afectan a uno de los 3 genes que codifican la proteína laminina 5: LAMA3, LAM3 o LAMC2, LAMB3, por lo que los hemidesmosomas de la membrana basal serían defectuosos. También se han descrito mutaciones en otros componentes de la lámina lúcida, como el colágeno XVII o la integrina α6β4; en contraste todas las formas de EB de unión son de herencia recesiva.5, 8
• EB Distrófica: La ampolla se localiza por debajo de la membrana basal a nivel de las fibrillas de anclaje. Al cicatrizar las sucesivas heridas crean retracción en las articulaciones afectando a la movilidad. Puede aparecer también en membranas mucosas: boca (puede restringir su apertura), faringe, esófago (dificulta para tragar incluso saliva), estómago, intestinos, vías respiratorias, urinarias, en las córneas (las heridas pueden producir pérdida de visión) y párpados. Estas formas distróficas son debidas a mutaciones en el gen del colágeno tipo 7, COL7A1. Son 10 genes bien identificados los que provocan este grupo de enfermedades y todos ellos producen proteínas de ensamblaje en la unión dermoepidérmica. Según el tipo de mutación resultante y su localización en la cadena de colágeno, las fibrillas de anclaje estarían defectuosas, disminuidas en cantidad y/o calidad, o ausentes. Recientemente se publicó una nueva forma letal epidérmica acantolítica de EB en un recién nacido por mutaciones en la desmoplaquina de los desmosomas.3, 7-8
La mayoría de las complicaciones de este grupo heterogéneo de enfermedades vienen determinadas por los deficientes mecanismos de reparación de las ampollas y por la localización de las mismas. En un extremo estarían las formas simples, con escasas complicaciones y en el otro, las formas distróficas, gravemente mutilantes e invalidantes.9
La sobre infección bacteriana es la principal complicación de todas las formas de EB. Las ampollas constituyen un caldo de cultivo excelente para microorganismos, sobre todo bacterias, que son capaces de ensombrecer el pronóstico estético, ya que determinan una cicatrización mucho más anómala e incluso a gravar el pronóstico vital por el riesgo de septicemia (la vía de entrada son las mucosas lesionadas). La realización de una exploración cuidadosa, atendiendo al aspecto de las lesiones, su olor, presencia de dolor, febrícula o fiebre, permitirán la sospecha de infección, siendo necesario en ocasiones medidas de asilamiento y la administración de antibióticos por vía sistémicas.9-10
El diagnóstico de las EB se realiza en base a aspectos clínicos, histopatológicos, biopatológicos y genéticos. También se deben contemplar las diferentes pruebas, disponibles hoy día, para un diagnóstico prenatal (DPN) de la enfermedad. El DPN es posible en la forma fetal recesiva o tipo Herlitz mediante la fetoscopía y la obtención de una biopsia de piel para estudio en microscopía electrónica y/o convencional apreciándose una separación entre la epidermis y la dermis a nivel de la lámina lúcida, junto con un número reducido de hemidesmosomas malformados. El tipo dominante distrófico se ha diagnosticado también in útero al detectar niveles elevados de alfa fetoproteína en el suero materno y en el líquido amniótico. El desarrollo de la Biología Molecular con las técnicas para el estudio de marcadores de ADN ha ampliado las posibilidades en el estudio y diagnóstico prenatal de muchas enfermedades, entre ellas, la epidermolisis bulosa.9-11
El diagnóstico diferencial debe hacerse con todas las dermatosis del recién nacido que cursan con ampollas y erosiones. En el examen clínico, hay que considerar la localización de las lesiones, la afectación de las mucosas, la existencia de infecciones y la posibilidad de otra sintomatología.En un primer momento se deben descartar las causas benignas más frecuentes, como serían la formación de ampollas por succión, ampollas por medicamentos, productos químicos, o quemadura por fototerapia de la ictericia neonatal. (12) El diagnóstico diferencial con otras patologías incluye:
• Síndrome de piel escaldada por estaphylocoucus (Enfermedad de Ritter).
• Herpes neonatal. La disposición en «ramillete» de las vesículas es una buena clave diagnóstica, si bien éstas pueden ser generalizadas (Dermatitis herpetiforme ampollar).
• Sífilis congénita. En la actualidad es muy rara. Cursa con ampollas palmo-plantares. Se deberá realizar serología (VDRL) a la madre y neonato, en este caso ambos resultados fueron negativos.
• Dermatosis autoimnunes. Son excepcionales. La más frecuente es el penfigoide del recién nacido por paso a la sangre del feto, por vía transplacentaria de anticuerpos de la madre.
• Otras patologías más infrecuentes como: mastocitosis, histiocitosis de Hashimoto, incontinencia pigmenti, eritrodermia ictiosiforme bulosa congénita, acrodermatitis bulosa, acrodermatitis enteropática y porfiria eritropoyética congénita.13
El objetivo final del tratamiento es ayudar a los pacientes afectados a vivir en la forma más completa y plena posible y es específico para cada paciente, ya que la severidad de la EB es muy variable. Dentro de las normas generales se incluye evitar los roces que causan las heridas, tratar las ampollas, vendar con apósitos especiales las lesiones y curarlas diariamente para evitar infecciones. También se deben tratar otros problemas comunes, como la anemia y el déficit de vitamina D. Hasta ahora la enfermedad no tiene tratamiento curativo, sólo sintomático y causa un severo daño físico, emocional y financiero al paciente y su familia.14
La educación de paciente, familiares y comunidad se considera fundamental para lograr el cumplimiento terapéutico, mantener los logros de los tratamientos médicos y de las intervenciones quirúrgicas, retrasar las recidivas, prevenir la pérdida de la funcionalidad y autonomía logradas, así como evitar una merma en la calidad de vida. La EB es una enfermedad poco frecuente y por ello, poco conocida, que supone un problema de gran magnitud en el entorno del enfermo. El apoyo social no evitará que estos pacientes sufran la enfermedad, pero puede ayudar a minimizar algunos de sus problemas, simplemente colaborando con los esfuerzos que se realizan para mejorar la información y coordinación.7, 15
REFERENCIAS BIBLIOGRÁFICAS
1. Miranda Gómez A, Frías Ancona G, Hierro Orozco S. Epidermólisis ampollosa. Revisión clínica. Revista Mexicana de Pediatría. [Internet]. 2003 [Citado 31 Mar 2010]; 70(1): [Aprox. 5p.]. Disponible en: http://www.medigraphic.com/pdfs/pediat/sp-2003/sp031h.pdf
2. Cordero MC, González BS, Castillo AC, Morales CE, Misad SC, Ruiz EF. Epidermólisis bulosa distrófica recesiva. Caso clínico. Rev Méd Chile. [Internet]. 2004[Citado 31 Mar 2010]; 132(5): [Aprox. 4p.]. Disponible en: http://www.scielo.cl/scielo.php?pid=s0034 -98872004000500012&script=sci_arttext
3. Balleste López I, Campo González A, Reyes Degournay R, Sanfiel Ferrer A. Epidermolisis bulosa, a propósito de un caso. Rev Cubana Pediatr. [Internet]. 2008 [Citado 31 Mar 2010]; 80(1): [Aprox. 8p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034 -75312008000100014&lng=es&nrm=iso&tlng=es
4. Ruiz Villaverde R, Blasco Melguizo J, Sánchez Cano D, Viciana Martínez L. Epidermólisis ampollosa juntural (tipo Herlitz). Med Cutan Iber Lat Am. [Internet]. 2006 [Citado 31 Mar 2010]; 34(5): [aprox. 2p.]. Disponible en: http://www.medigraphic.com/pdfs/cutanea/mc-2006/mc065h.pdf
5. Fine JD, Eady RAJ, Bauer JA, Bauer JW , Bruckner-Tuderman L , Heagerty A , et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. [Internet]. 2008 [Citado 31 Mar 2010]; 58(6): [Aprox. 19p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18374450
6. Baquero Fernández C, Herrera Ceballos E, López Gutiérrez JC, De Lucas Laguna R, Romero Gómez J, Serrano Martínez MC. Guía de atención clínica integral de la epidermólisis bulosa hereditaria. [Internet]. Madrid; 2008 [citado 31 mar 2010]. Disponible en: http://www.msc.es/profesionales/prestacionesSanitarias/publicaciones/docs/epidermolisisBullosa.pdf
7. Weinel S, Lucky AW, Uitto J, Pfendner EG, Choo D. Dystrophic epidermolysis bullosa with one dominant and one recessive mutation of the COL7A1 gene in a child with deafness. Pediatr Dermatol. [Internet]. 2008 [Citado 31 Mar 2010]; 25(2): [Aprox. 4p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18429782
8. Fine JD, Mellerio J. Extracutaneous manifestations and complications of inherited epidermolysis bullosa. Epithelial associated tissues.J Am Acad Dermatol. [Internet]. 2009 [Citado 31 Mar 2010]; 61(3): [Aprox. 17p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19700010
9. Narendran V, Steven B. Epidermolysis Bullosa. En: Martin RJ, Fanaroff AA, Walsh MC. Fanaroff and Martin's Neonatal-Perinatal Medicine. Diseases of the Fetus and Infant. Philadelphia: Mosby, An Imprint of Elsevier; 2006. [Aprox. 4p.] Disponible en: http://www.amazon.com/Fanaroff-Martins-Neonatal-Perinatal-Medicine-Diseases/dp/0323029663
10. Yiasemides E, Walton J, Marr P, Villanueva EV, Murrell DF. A comparative study between transmission electron microscopy and immunofluorescence mapping in the diagnosis of epidermolysis bullosa. Am J Dermatopathol. [Internet]. 2006 [Citado 31 Mar 2010]; 28(5): [Aprox. 7p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/17012912
11. Yancey KB. The Pathophysiology of autoinmune blistering diseases. Journal of Clinical Investigation. [Internet]. 2005 [Citado 31 Mar 2010]; 115(4): [Aprox. 5p.]. Disponible en: http://www.jci.org/articles/view/24855
12. Valdivielso M, Hernanz JM. Epidermólisis ampollosa simple hereditaria. Acta Pediatr Esp. 2008; 66:25-6.
13. Salas JC. Las epidermolisis bulosas. Med Cutan Iber Lat Am. [Internet]. 2007 [Citado 31 Mar 2010]; 35(4): [Aprox. 2p.]. Disponible en: http://www.medcutan-ila.org/articulos/2007/4/pdf/07-034.pdf
14. Fine JD, Hintner H. Life with Epidermolysis Bullosa: Etiology, Diagnosis, and Multidisciplinary Care and Therapy Wien. New York: Springer Verlag GmbH; 2009. Disponible en: http://www.springer.com/medicine/dermatology/book/978-3-211-79270-4
Recibido: 9 de noviembre de 2011.
Aprobado: 4 de octubre de 2012.

{kind=link}