










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.16 no.6 Pinar del Río nov.-dic. 2012
ARTÍCULO REVISIÓN
Aspectos moleculares y asesoramiento genético de los síndromes de sobrecrecimiento
Molecular aspects and genetic counseling of overgrowth syndromes
Reinaldo Menéndez García1, Anitery Travieso Téllez2, Miladys Orraca Castillo3, Deysi Licourt Otero4
1Especialista de Segundo Grado en Genética Clínica. Profesor Auxiliar. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico:generey@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral y Genética Clínica. Centro Provincial de Genética Médica. Pinar del Río, Cuba. Correo electrónico:any0511@princesa.pri.sld.cu
3Especialista de Segundo Grado en Genética Clínica. Profesora Auxiliar. Máster en Atención Integral al Niño. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico:milgene@princesa.pri.sld.cu
4Especialista de Primer Grado en Medicina General Integral y de Segundo Grado en Genética Clínica. Asistente. Máster en Atención Integral al Niño. Investigadora Agregada. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico:deysili@princesa.pri.sld.cu
RESUMEN
El crecimiento y desarrollo constituyen dos conjuntos de elementos de gran utilidad para determinar el estado de salud de los pacientes en edad pediátrica. Actualmente se tiene constancia de numerosos trastornos de hipercrecimiento, habiéndose identificado un nutrido número de factores causantes, tanto genéticos como hormonales, pero hasta la fecha se han enunciado solo los mecanismos patogénicos implicados en una minoría de éstos, ya que la etiopatogenia y bases moleculares de las anomalías que cursan con hipercrecimiento son muy complejas y parcialmente conocidas, a pesar de que este trastorno resulta relativamente frecuente en nuestra población. En este trabajo se profundiza en el conocimiento de los aspectos moleculares asociados con la génesis de los síndromes de sobrecrecimiento de origen genético y los procesos de asesoramiento genético necesarios en el manejo de pacientes y familiares además de explicar los mecanismos bioquímicos que participan en su origen para fundamentar la necesidad de un Asesoramiento Genético individualizado dirigido a pacientes y familiares afectados por el síndrome.
DeCS: Crecimiento/genética, Desarrollo infantil.
ABSTRACT
Growth and development constitute two sets of very useful elements to determine the patients' health status in pediatric ages. Currently, there are proofs of numerous disorders of overgrowth, where a full number of leading factors have been identified both genetic and hormonal, up to the present, only the pathogenic mechanisms involved in a minority of these cases have been stated, since the etiology and pathogenesis plus the molecular basis of the anomalies coursing with overgrowth are very complex and partially known. Despite this disorder is relatively frequent in the population, this research deepened in knowledge regarding the molecular aspects associated with the genesis of overgrowth syndromes, the genetic origin as well as the process of the necessary genetic counseling to treat patients and their families; explaining at the same time the biochemical mechanisms involve in its origin in order to back up the need of Individualized Genetic Counseling to patients and families suffering from this syndrome.
DeCS: Growth/genetics, Child development.
INTRODUCCIÓN
El crecimiento en el ser humano es un proceso sumamente complejo ya que depende de múltiples factores genéticos, hormonales y am bientales, y todos ellos juegan un papel relevante. Las anomalías caracterizadas por hipercrecimiento o velocidad de crecimiento excesiva es una condición en la que hay excesivo crecimiento o desarrollo, localizado o generalizado para la edad y sexo del individuo. Los diversos estudios y observaciones ilustran esta complejidad de este proceso y el amplio número de genes y factores reguladores que intervienen en el desarrollo de un crecimiento normal proporcionado.1
El sobrecrecimiento puede ser generalizado cuando la mayoría o todas las variables antropométricas mensurables, de crecimiento y desarrollo, están aumentadas y se considera localizado o regional, cuando afecta sólo a una parte o varias regiones del cuerpo.1
Hasta ahora se tenía constancia de numerosos trastornos de hipercrecimiento, habiéndose identificado un número de factores causantes, tanto genéticos como hormonales, pero los mecanismos patogénicos implicados se han enunciado solo en una minoría de estos, ya que la etiopatogenia y bases moleculares de las anomalías que cursan con hipercrecimiento son muy complejas y parcialmente desconocidas.2
Los síndromes de sobrecrecimiento, aunque raros, son diagnosticados con mayor frecuencia de manera tardía. Los progresos en la identificación de sus causas genéticas, han permitido conocer sus manifestaciones más y menos frecuentes y establecer la correlación genotipo-fenotipo, así como conocer los principales mecanismos fisiopatológicos.3 El sobrecrecimiento congénito es definido cuando el peso neonatal está por encima del 97 percentil, mientras que en la niñez y la adultez está presente, si la relación talla/peso se encuentra por encima del 97 percentil.4
El sobrecrecimiento mayormente es resultante de los siguientes procesos:
a) Incremento del número de células o hiperplasia.
b) Hipertrofia.
c) Incremento del intersticio, fundamentalmente el líquido intersticial.
d) Alguna combinación de los factores anteriores.5
Teniendo en cuenta que este trastorno resulta relativamente poco conocido se decide profundizar en los aspectos moleculares relacionados con la etiología de alguno de los síndromes de hipercrecimiento, así como fundamentar la necesidad de un adecuado proceso de asesoramiento genético para el seguimiento y el manejo integral de las familias con miembros afectados.
DESARROLLO
El crecimiento pre y postnatal se mueve dentro de un amplio rango de valores considerados dentro de la normalidad. Las variables antropométricas más utilizadas en la práctica clínica pediátrica son la medición del peso, medición talla y del perímetro cefálico.
Los niños con déficit o retraso del crecimiento crecen con dos o más de los tres parámetros por debajo del percentil tercero, mientras que el sobrecrecimiento ("macrosomía", "bebé macrosómico", "gigante", "grande para la edad gestacional") es una condición en la que hay excesivo crecimiento o desarrollo, localizado o generalizado para la edad y sexo del individuo.2
El crecimiento pre y postnatal tiene, como cualquier parámetro biológico, un grado de variabilidad notable, existiendo un amplio intervalo de valores considerados dentro de la normalidad. Las variables antropométricas preferentemente utilizadas en la práctica clínica son la medición del peso, talla y perímetro cefálico. El sobrecrecimiento o hipercrecimiento se puede definir por valores, de cualquiera de los anteriores parámetros (talla, peso y perímetro cefálico), situados dos desviaciones estándar (DE) por encima de la media o más, para la población de referencia o por encima del percentil 95 para la edad y sexo correspondiente.2,3
El 5% de la población de niños recién nacidos tendrá alguno de estos parámetros por encima del percentil 95 (+ 2 desviaciones estándar). Esta probabilidad es del 0,25 %, es decir de 1/400. Para una adecuada orientación diagnóstica se precisa de la aplicación sistemática y exhaustiva del método clínico que permite la aproximación a una de las variantes de sobrecrecimiento, para lo que se debe descartar primero aquellas más frecuentes dentro de la población.
Para ello se ha de tener en cuenta la clasificación según etiología probable de la talla alta.6
Clasificación:
Variantes normales.
· Talla alta normal.
· Maduración acelerada familiar.
II. Obesidad.
III. Causas hormonales.
IV. Talla alta sindrómica.
IV.1. Trastornos cromosómicos.
• Síndrome de Klinefelter.
• Síndromes 47 XXX, 47 XYY.
• Trisomía 8, mosaicismo.
• Trisomía 8p.
IV. 2. Otros síndromes genéticos.
• Síndromes Marfanoides.
• Síndrome Partington.
• Neurofibromatosis 1.
• Síndrome Teebi.
• Síndrome frágil X.
IV.3. Hipercrecimiento de comienzo prenatal.
• Síndrome Beckwith-Wiedemann.
• Síndrome Sotos.
• Síndrome Weaver.
• Síndrome Simpson-Golabi-Behmel.
• Síndrome Nevo.
• Síndrome Elejalde.
• Síndrome Perlman.
Síndrome Marshall Smith.
• Síndrome Bannayan-Riley-Ruvalcaba.
Los síndromes de sobrecrecimiento se acompañan por un patrón de signos dismórficos que se asocian, siendo la alta talla la característica relevante.5 Los pacientes que los padecen tiene un mayor riesgo de retraso mental (leve, moderado o grave) que la población general (1-3 % versus 10 %) y un mayor riesgo de desarrollar tumores en edad pediátrica (1:14.000.versus el 3 al 40 % en dependencia del síndrome).7
Un hecho sobresaliente de los síndromes de sobrecrecimiento es el riesgo de cáncer. Aunque en los años precedentes ha habido algunas recomendaciones para el screening tumoral en algunos de estos síndromes, éstas son empíricas y no específicas; por lo tanto, no hay protocolos específicos para todos y cada uno de ellos. En algunos de ellos (Perlman, Beckwith Wiedemann, Simpson-Golabi-Behmel y hemihipertrofia), los tumores aparecen principalmente en el abdomen (más del 94 % de los tumores), generalmente antes de los 10 años y son, principalmente, embrionarios. Por el contrario, en otros, como el Sotos, los tumores más frecuentes fueron los de tipo linfo-hematológicos, entre el 60-70 % son extra abdominales y un gran número aparecen después de los 10 años.7, 8
Aunque raras, estas entidades son diagnosticables en la población y esta realidad se asocia a que cada vez resultan más los trastornos genéticos identificados con el hipercrecimiento y es mayor el progreso del conocimiento sobre los aspectos moleculares relacionados con su etiopatogenia, sus mecanismos fisiopatológicos y la correlación genotipo-fenotipo en los individuos afectados.9
Existe una gran diversidad y heterogeneidad de características asociadas a los distintos trastornos y síndromes de hipercrecimiento, por lo que es difícil hacer una clasificación de distintos rasgos comunes asociados a estas afecciones. Sin embargo, los diferentes cuadros de hipercrecimiento generalizado en el nacimiento, suelen compartir algunas características, que incluyen:
Peso con incremento proporcional al aumento de la altura.
El sobrecrecimiento acompañando otras anomalías.
Asociación con frecuencia a cierto grado de retraso mental.
Riesgo de neoplasias en diferentes tejidos (Ej.: tumor de Wilms, leucemia, neuroblastoma y astrocitoma).10
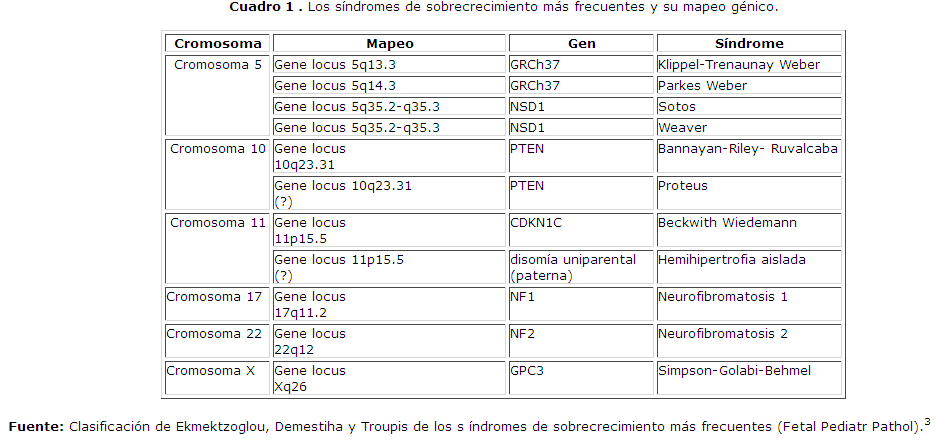
Se reflejan los síndromes de sobrecrecimiento más frecuentes, el gen responsable que lo produce, su ubicación cromosómica y el locus específico donde se encuentra mapeado, cuadro 1 y cuadro 2.
A continuación, se analizan los más recientes aportes de la biología molecular al origen de la alta talla sindrómica, recorriendo aquellas entidades más frecuentes en las consultas de Genética Clínica.11-13
Síndrome Marfán
El síndrome de Marfán consiste en una alteración del tejido conectivo que afecta al esqueleto humano, con elongación de los huesos tubulares, y alteraciones en el sistema cardiovascular y el sistema ocular. Los pacientes afectos de este síndrome suelen presentar además moderada laxitud articular, dedos y extremidades desproporcionadamente largos, dientes apiñados, escoliosis y lordosis torácica. (Figura 1)La esperanza de vida de estos pacientes suele estar mermada por complicaciones cardiovasculares, ya que suelen presentar debilidad de la túnica media de los grandes vasos originando una dilatación de la aorta ascendente o arteria pulmonar y/o aneurisma disecante, con frecuencia pueden presentar también prolapso de la válvula mitral.15
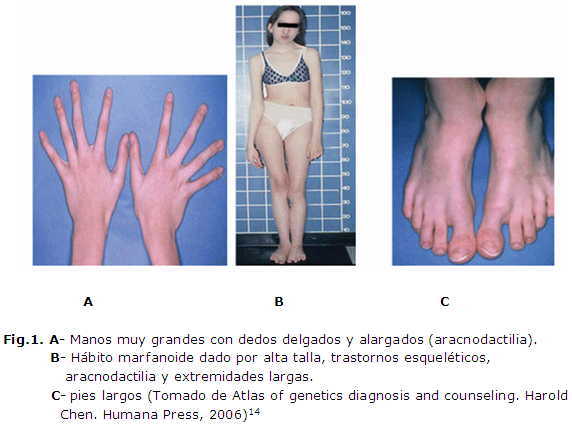
Este trastorno con patrón de herencia autosómico dominante es provocado por mutaciones en el gen FBN1 ubicado en 15q21, la mayoría de ellas mutaciones puntuales habiéndose encontrado pocos casos de grandes reordenamientos afectando a este gen. El síndrome Marfán es una fibrilinopatía originada por una inadecuada síntesis de la proteína fibrilina 1 a raíz de alteraciones del gen FBN1. La fibrilina es el componente más importante del tejido conectivo y muy probablemente un elemento funcional esencial, por lo que la producción de proteína alterada explica los signos clínicos y los cambios histológicos observados en los pacientes con mutaciones en el gen FBN1 al interferir en la organización normal de las microfibrillas.14,15
Se ha definido un síndrome de Marfán tipo II o Loeys Dietz por el hallazgo de mutaciones en los genes TGFBR1 (9q33) y TGFBR2 (3p22) en algunos pacientes en los que previamente no se habían encontrado mutaciones afectando al gen FBN1, estos pacientes presentan un fenotipo bastante similar al producido por el síndrome Marfán clásico, aunque no presentan anomalías oculares y su esperanza de vida parece ser menor.15-17
Síndrome de Beckwith-Wiedemann
El Síndrome de Beckwith-Wiedemann es el más común de los síndromes de sobrecrecimiento; caracterizado por macrosomía, macroglosia, organomegalia y anomalías del desarrollo (en particular los defectos de la pared abdominal con exónfalos/onfalocele) 18. También pueden padecer durante la infancia hemihiperplasia e hipoglicemia, así como un riesgo incrementado de sufrir tumores calculado entre el 5 % y el 10 %. La incidencia de este síndrome está estimada en 1 de cada 14.000 nacimientos.19 (Figura 2 )

Aunque la mayoría de los casos de Beckwith-Wiedemann son esporádicos 85 %, este es un desorden complejo, multigénico asociado en más del 90% de los pacientes con una alteración en la expresión o función de uno o más genes en la zona 11p15.520. Alrededor del 15 % se corresponden con formas familiares.
La región 11p15.5 incluye genes que codifican factores de crecimiento y genes supresores de tumores, varios de los genes reguladores están sometidos a impronta genómica. Los genes de expresión paterna (impronta materna) tienen actividad potenciadora del crecimiento y los de expresión materna (impronta paterna) tienen actividad supresora del crecimiento.16
La región 11p15 tiene dos dominios diferentes de impronta, uno telomérico que contiene los genes H19 e IGF2 y otro centromérico que incluye: KCNQ1, KCNQ10T1 (LIT1) y CDKN1C. Se han descrito mutaciones puntuales intragénicas en el gen CDKN1C, microdeleciones de LIT1, alteraciones de la metilación en el gen H19, duplicaciones de origen paterno, translocaciones cromosómicas de origen materno y otras. Es por ello, que tanto mecanismos cromosómicos, genéticos o epigenéticos pueden darse en pacientes con este síndrome.2, 19
Una serie de estudios recientes sugieren que las técnicas de reproducción asistida pueden incrementar los riesgos de defectos de la impronta, y en particular del síndrome de Beckwith-Wiedemann, similares observaciones se han reportado entorno al síndrome de Angelman. En ambos trastornos después de la reproducción asistida la alteración observada siempre implica la pérdida de metilación de los genes metilados de impronta materna, lo que sugiere que las técnicas de reproducción asistida impiden la adquisición o mantenimiento de las marcas de metilación en los genes de impronta materna.21, 22
Síndrome Sotos
El síndrome Sotos fue descrito en 1964, está conformado por talla alta, gran dolicocefalia, configuración facial característica (100 %), edad ósea avanzada (84 %) y anomalías neurológicas no progresivas con retraso mental. (Figura 3 ). La prevalencia es desconocida, aunque se trata, probablemente, de uno de los síndromes más frecuentes de hipercrecimiento, después del síndrome de Marfán y el síndrome de Williams-Beuren.3, 23
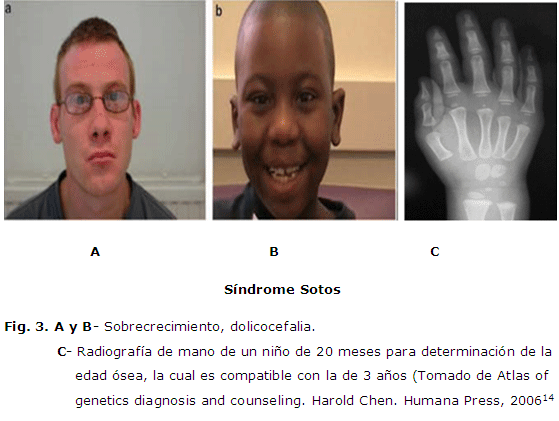
El hallazgo clínico fundamental es el hipercrecimiento prenatal y postnatal. La talla adulta en varones se sitúa en torno a 184,3 ± 6,0 cm y en mujeres en torno a 172,9 ± 5,7 cm, lo que supone un incremento respecto de la talla genética de 11 y 6 cm, respectivamente. No obstante, se han descrito tallas adultas más elevadas: de 1.93 cm a 2.03 cm en varones y hasta 1.88 cm en mujeres.
La configuración craneofacial es muy característica: frente prominente, disminución de la línea del cabello frontopariental, gran dolicocefalia, hipertelorismo, inclinación antimongoloide de los párpados, paladar ojival y estrecho, palatino prominente serrado y mentón prominente. Aproximadamente, el 60-80 % de los pacientes presentan una erupción prematura de sus dientes. Las manifestaciones del sistema nervioso central son frecuentes. El retraso en las adquisiciones del desarrollo, para caminar, hablar y, en particular el lenguaje, está casi siempre presente y la torpeza es frecuente (60-80 % de los casos), como lo es la hipotonía y la laxitud articular.
La deficiencia mental está presente en el 80-85 % de los casos, exhibiendo un coeficiente intelectual de 72 y un rango entre 40 y límite de la normalidad. Las convulsiones pueden estar presentes en un 30 % de los casos. Existe una incidencia incrementada de tumores cerebrales (2,2-3,9 %).
La presencia de ventrículos cerebrales ligeramente alargados y la presencia de espacios subaracnoideos aumentados, son hallazgos frecuentes que, habitualmente, no requieren ningún tipo de tratamiento. Otras anomalías que pueden presentarse, son las siguientes: defectos septales cardíacos, cifoescoliosis, alteraciones urogenitales (hidronefrosis, riñones hipoplásicos, agenesia renal, dilatación de la pelvis renal), hernias inguinales, alteraciones oftalmológicas (estrabismo, anomalías del nervio óptico o de la retina), alteraciones endocrinológicas (hipotiroidismo primario, entre otras). No obstante, no se han descrito alteraciones endocrinológicas capaces de explicar el rápido crecimiento de estos pacientes.23
La existencia de una causa genética se ha sospechado desde hace mucho tiempo, habiéndose descrito varias familias con miembros afectos en dos o tres generaciones, sugiriendo un patrón de herencia autosómico dominante. No obstante, la mayoría de los casos son esporádicos, aunque podrían ser debidos a nuevas mutaciones (afectando preferentemente al cromosoma paterno). Recientemente, se han descrito mutaciones en el gen NSD1 (5q35) en el 60-75 % de casos esporádicos, indicando que la haploinsuficiencia del gen NSD1 es la causa más importante de síndrome de Sotos.14, 24 No obstante, es posible que existan otras anomalías genéticas.
El gen humano NSD1 consta de 23 exones, codifica una proteína de 2696 aminoácidos y se expresa en el cerebro fetal humano, lo que podría explicar el gran cerebro y la deficiencia mental en estos pacientes. También se expresa en el músculo esquelético, riñón, bazo, leucocitos periféricos y timo. La proteína a la que da lugar este gen interactúa con un dominio de unión a receptores hormonales nucleares, pudiendo actuar como co-represor o como co-activador. El hallazgo de que la haploinsuficiencia del gen NSD1 induce hipercrecimiento en el síndrome de Sotos, implica que el gen NSD1 actúa como co-represor de genes que promueven el crecimiento. Todos los ratones "knock-out" homocigotos para el gen NSD1 fallecen antes del día embrionario 10,5. Por consiguiente, se considera que el gen NSD1 es crucial para el desarrollo postimplantación. La infertilidad, el aborto y los niños nacidos muertos, podrían ser una explicación para el bajo número de casos familiares.14
No existe ningún marcador bioquímico para este síndrome. El diagnóstico se basa, preferentemente, en sus aspectos clínicos. Las manifestaciones clínicas más características son la configuración cráneo-facial y el crecimiento excesivo. Sin estos datos, no puede efectuarse el diagnóstico.24, 25
Síndrome de Weaver
El Síndrome de Weaver fue descrito en 1974. Se caracteriza por hipercrecimiento prenatal y postnatal, facies inusual, maduración ósea avanzada y camptodactilia. Se han comunicado al menos 37 casos (21 varones y 16 mujeres). Los varones alcanzan una talla adulta en torno a 194,2 cm, peso de 102,2 kg y un perímetro craneal de 61 cm. Las mujeres alcanzan una talla adulta alrededor de 176, 3 cm, un peso de 87, 6 kg y un perímetro craneal de 59, 5 cm. El hipercrecimiento está presente al nacimiento o se inicia durante la lactancia. Aproximadamente el 80 % de los pacientes están retrasados desde el punto de vista del desarrollo o tienen retraso mental. El retraso mental puede ser desde ligero hasta severo; muchos de estos elementos se asemejan al síndrome de Sotos. (Figura 4 )
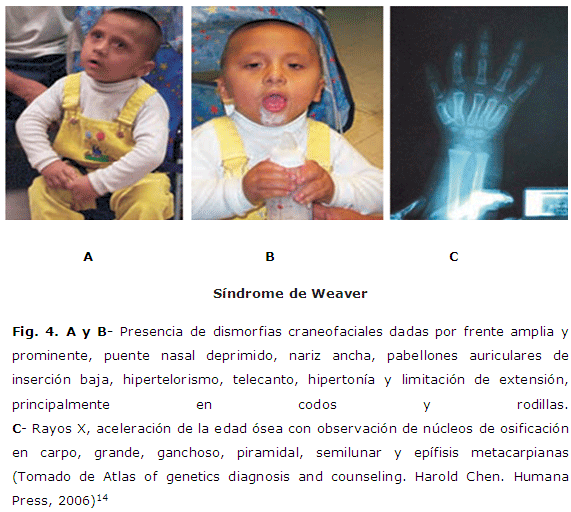
Las características cráneo faciales, aunque algo similares, son algo diferentes. En efecto, los pacientes con síndrome de Weaver presentan hipertelorismo, pabellones auriculares grandes, puente nasal deprimido, inclinación antimongoloide de los párpados, pero el cráneo, en muchos casos, no es dolicocéfalo (el occipucio es, a menudo, plano) y no tienen mentón prominente y la cara es ancha.26
Otro elemento característico es que, en muchos casos, existe hipertonía más que hipotonía. Es muy llamativa la existencia de una maduración ósea avanzada, con una madurez mucho más acelerada en el carpo que en la mano. Los huesos largos están ensanchados o en bisel, y la camptodactilia es frecuente. La mayoría de los casos son esporádicos, si bien se ha comunicado transmisión de padres a hijos en cinco ocasiones, sugiriendo un patrón de herencia autosómico dominante.26, 27
Recientemente, se han descrito mutaciones en el gen NSD1 en el 46 % de pacientes (6 de 13) con síndrome de Weaver, lo que sugiere que el síndrome de Sotos y el síndrome de Weaver son síndromes alélicos. La mayor parte de los individuos con este diagnóstico resultan heterocigóticos para las mutaciones del gen NSD1.27, 28
Asesoramiento genético de los síndromes de sobrecrecimiento.
La Sociedad Americana de Genética Humana, ha definido el asesoramiento genético como un proceso de comunicación relacionado con problemas humanos que se generan con la ocurrencia de una enfermedad genética en una familia o sus riesgos de recurrencia y que lleva implícita la intervención de una o más personas capacitadas, para ayudar al paciente y su familia a comprender los hechos médicos, incluyendo el diagnóstico, la historia natural de la enfermedad y atención o tratamiento posible; entender los mecanismos genéticos por los cuales se produce el padecimiento y el riesgo de recurrencia en familiares específicos, conocer diversas opciones para manejar el riesgo de recurrencia, elegir el curso de la opción que los pacientes consideren apropiado de acuerdo a sus riesgos y metas familiares, actuar de acuerdo a esa decisión, así como realizar la mejor adaptación posible a la presencia de un miembro afectado y un riesgo determinado.29-30 Como se ha expuesto el diagnóstico clínico de pacientes con hipercrecimiento puede ser complicado, dada la gran variabilidad fenotípica de este síndrome y la existencia de numerosos trastornos que cursan con alta talla y otras características clínicas solapantes.
Generalmente el manejo de los síndromes de sobrecrecimiento incluye una evaluación inicial en el momento del diagnóstico donde se establece la magnitud o grado de compromiso de la enfermedad, tratamiento de las manifestaciones clínicas, la prevención de complicaciones secundarias y se establece el pronóstico de supervivencia y calidad de vida. Su tratamiento generalmente incluye a un equipo multidisciplinario y es adecuado que así sea donde participan genetistas clínicos, pediatras, internistas, angiólogos, ortopédicos, radiólogos, cardiólogos, oftalmólogos, psicoterapeutas, entre otros.
El diagnóstico diferencial debería incluir, al menos, el síndrome de Weaver, el síndrome de Beckwith-Wiedemann y el síndrome de Sotos ya que pueden ser los trastornos de hipercrecimiento que más hallazgos clínicos comparten, además del sobrecrecimiento pre y postnatal, incluyen la hipotonía neonatal, edad ósea adelantada, anomalías cardíacas y riesgo de neoplasias entre otros. El manejo de estos pacientes debe comenzar desde el propio pediatra o médico de familia, con la derivación a un clínico especialista tras la observación de signos y hallazgos clínicos sugerentes de hipercrecimiento o de trastorno del desarrollo.
Realizar un examen físico completo, mesurando parámetros físicos de crecimiento, realizando un examen renal, cardíaco y ortopédico. En el caso de encontrar anomalías en estos estudios, referir el paciente al especialista apropiado para su adecuado tratamiento. También es importante la realización de test para medir coeficiente de inteligencia, ya que estos pacientes suelen presentar diferentes grados de discapacidad intelectual.1-3, 14, 27
Una vez que el genetista clínico ha establecido un diagnóstico a punto de partida de los hallazgos clínicos el siguiente paso es llevar a cabo un adecuado proceso de asesoramiento genético al paciente y su familia. Ello prepara a uno y otros para aceptar el trastorno y aprender a vivir con él, así como orienta sobre el riesgo de recurrencia en la familia y las medidas a tomar prevenir complicaciones.29
Los síndromes de hipercrecimiento descritos coinciden en que la mayoría presentan un modelo clásico de herencia autosómica dominante por lo que el individuo afectado tiene un 50% de riesgo en cada embarazo de transmitir la enfermedad a sus descendientes sin distinción de sexo. Cuando ocurre un primer caso en la familia interpretado como una nueva mutación los padres no presentan un alto riesgo.
Luego de la revisión de los aspectos moleculares de los síndromes de sobrecrecimiento y el comportamiento fenotípico, el autor de esta revisión sugiere una metodología para el manejo de los pacientes que debe comenzar por establecer su correcto diagnóstico el cual se realiza iniciando con un examen físico completo, midiendo parámetros físicos de crecimiento, realizando un examen renal, cardiovascular, ortopédico y un test para determinar el coeficiente de inteligencia. En resumen, la conducta a seguir con un paciente afectado de síndrome de sobrecrecimiento son las que aparecen en el manual de normas y procederse para la Genética Clínica, cuyos aspectos esenciales son los siguientes:
1. Confeccionar la historia clínica completa que incluya el árbol genealógico.
2. Realizar dermatoglifos al paciente y a ambos padres.
3. Determinar cromatina sexual en mucosa oral.
4. Cariotipo en sangre periférica al paciente.
5. Test inmunohistoquímico o estudio molecular para descartar Síndrome Frágil X según criterios clínicos.
6. Imágenes neurológicas: (TAC, RMN y ultrasonido transfontanelar).
7. Ecografía abdominal, renal y cardiaca.
8. Rayos X de manos para determinación de la edad ósea.
9. Interconsultas obligatorias con Endocrinología, Fisiatría, Logofoniatría, y Neuropediatría.
10. Interconsultas si fueran necesarias con Otorrinonaringología, Ortopedia, Oftalmología, Cardiología y Oncopediatría.
11. Realizar complementarios: hemoglobina, hematocrito, leucograma, eritrosedimentación, transaminasa para estudio de la función hepática, parcial de orina, glicemia y prueba de tolerancia a la glucosa (PTG).
12. Seguimiento anual en la Consulta de Genética Clínica.
CONCLUSIONES
El seguimiento del plan anterior es beneficioso porque permite el asesoramiento genético a los padres, brinda orientaciones encaminadas a prevenir complicaciones asociadas, favorece y guía la intervención multidisciplinaria, mejora la calidad de vida del paciente y sus familiares y facilitauna mejor integración del paciente a la sociedad.
Se debe llevar a cabo un adecuado proceso de asesoramiento genético al paciente y su familia es muy importante porque al presentar un modelo de herencia clásico autosómico dominante, el individuo afectado tiene un 50 % de riesgo en cada embarazo de transmitir la enfermedad a sus descendientes y es necesario orientarlos, informarles el riesgo que representa para su descendencia, ayudarlos a manejar las complicaciones y ayudarlos a tomar decisiones reproductivas responsables y conscientes.
REFERENCIAS BIBLIOGRÁFICAS
1. Sotos JF, Argente J. Overgrowth disorders associated with tall stature. Adv Pediatr. [Internet]. 2008 [citado 28 Mar 2012]; 55: [Aprox. 34p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19048732 [buscar en Scholar Google]
2. Thomsett MJ. Referrals for tall stature in children: a 25-year personal experience. J Paediatr Child Health. [Internet]. Ene-Feb 2009 [citado 28 Mar 2012]; 45(1-2): [Aprox. 6 p.]. Disponible en: http://dx.doi.org/10.1111/j.1440-1754.2008.01428.x
3. Ekmektzoglou K, Demestiha T, Troupis G, Xanthos T. Commonest Overgrowth Syndromes. Fetal Pediatr Pathol. [Internet]. Abr 2012 [citado 28 Mar 2012]; 31(2): [Aprox. 16p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22409408
4. Kant SG, Wit JM, Breuning MH. Genetic analysis of tall stature. Horm Res [Internet]. Sep 2005 [citado 28 Mar 2012]; 64(3): [Aprox. 8 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/16192740
5. Cohen MM, Neri G, Weksberg R. Overgrowth Syndromes. England: Oxford University Press; 2002.
7. Neri G, Moscarda M. Overgrowth syndromes: a classification. Endocr Dev [Internet]. 2009 [citado 28 Mar 2012]; 14: [aprox. 8 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19293574
8. Lapunzina P, Gracia Bouthelier R. Prevención y detección precoces de neoplasias en los síndromes de sobrecrecimiento. Pediatr Integra. 2006; 10(8).
9. Romanelli V. Análisis genético y correlaciones clínico-moleculares en pacientes con síndrome de sobrecrecimiento. [Tesis Doctoral]. España: Universidad Autónoma de Madrid; 2010 [citado 28 Mar 2012]. Disponible en: http://www.tdx.cat/handle/10803/50341
10. Chawarska K, Campbell D, Chen L, Shic F, Klin A, Chang J. Early generalized overgrowth in boys with autism. Arch Gen Psychiatry. [Internet]. 2011 [citado 28 Mar 2012]; 68(10): [Aprox. 10p.]. Disponible en: http://archpsyc.ama-assn.org/cgi/content/abstract/68/10/1021
11. Choufani S, Shuman C, Weksberg R. 2010. Beckwith_Wiedemann syndrome. Am J Med Genet Part C 153C:343-354.
12. Ruggieri VL, Arberas CL. Síndromes genéticos reconocibles en el periodo neonatal. Medicina (Buenos Aires) [Internet]. 2009 [citado 28 Mar 2012]; 69(1/1): [Aprox. 21p.]. Disponible en: http://www.scielo.org.ar/pdf/medba/v69n1s1/v69n1s1a04.pdf
13. Marín Gómez P, García García E, Lapunzina Badía P. Caracterización y Atención Temprana del Síndrome de Sotos. Psicología Educativa. [Internet]. 2011 [citado 28 Mar 2012]; 17(2): [Aprox. 16 p.]. Disponible en: http://www.copmadrid.org/webcopm/publicaciones/educativa/ed2011v17n2a7.pdf
14. Valle Domínguez JM. Caracterización molecular del síndrome de sotos y estudio de otras causas genéticas de hipercrecimiento. [Internet]. Universidad Pompeu Fabra; 2009 [citado 28 Mar 2012]. Disponible en: http://repositori.upf.edu/handle/10230/12274?show=full
15. Chen H. Atlas of genetic diagnosis and counseling: Silver-Russell Syndrome. Totowa: Human Press; 2006.
16. Chaffins JA. Marfan syndrome. Radiol Technol. [Internet]. 2007 [citado 28 Mar 2012]; 78(3): [Aprox. 14]. Disponible en: http://www.radiologictechnology.org/content/78/3/222.abstract
17. Bonetti MI . Microfibrils: a cornerstone of extracellular matrix and a key to understand Marfan syndrome. Ital J Anat Embryol. [Internet]. 2009 [citado 28 Mar 2012]; 114(4): [Aprox. 23p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20578676
18. Zangwill SD, Brown MD, Bryke CR, Cava JR, Segura AD. Marfan syndrome type II: there is more to Marfan syndrome than fibrillin 1. Congenital Heart Dis [Internet]. Sep-Oct 2006 [citado 28 Mar 2012]; 1(5): [aprox. 4 p.]. Disponible en: http://dx.doi.org/10.1111/j.1747-0803.2006.00040.x
19. Nativio R, Sparago A, Ito Y, Weksberg R, Riccio A, Murrell A. Disruption of genomic neighbourhood at the imprinted IGF2-H19 locus in Beckwith-Wiedemann syndrome and Silver_Russell syndrome. Hum Mol Genet [Internet]. 2011 [citado 28 Mar 2012]; 20(7): [Aprox. 12 p.]. Disponible en: http://hmg.oxfordjournals.org/content/20/7/1363.full.pdf+html
20. Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet [Internet]. 2010 [citado 28 Mar 2012]; 18(1): [aprox. 7 p.]. Disponible en: http://www.nature.com/ejhg/journal/v18/n1/pdf/ejhg2009106a.pdf
21. Romanelli V, Meneses HM, Fernández L, Martínez González V, Gracia-Bouthelier R, Fraga MF, et al. Beckwith_Wiedemann syndrome and uniparental disomy 11p: fine mapping of the recombination breakpoints and evaluation of several techniques. Eur J Hum Genet. [Internet]. 2011 [citado 28 Mar 2012]; 19 (4): [Aprox. 6p.]. Disponible en: http://www.nature.com/ejhg/journal/v19/n4/pdf/ejhg2010236a.pdf
22. Spivey PS, Bradshaw WT. Recognition and management of the infant with Beckwith-Wiedemann Syndrome. Adv Neonatal Care. [Internet]. 2009 [citado 28 Mar 2012]; 9(6): [Aprox. 5p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20010144
23. Bliek J, Verde G, Callaway J, Maas SM, De Crescenzo A, Sparago A, et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet [Internet]. 2009 [citado 28 Mar 2012]; 17(5): [Aprox. 9 p.]. Disponible en: http://www.nature.com/ejhg/journal/v17/n5/pdf/ejhg2008233a.pdf
24. Pasillas MP, Shah M, Kamps M. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Human Mutation. [Internet]. Mar 2011 [citado 28 Mar 2012]; 32(3): [Aprox. 7 p.]. Disponible en: http://dx.doi.org/10.1002/humu.21424
25. Cytrynbaum CS, Smith AC, Rubin T, Weksberg R. Advances in overgrowth syndromes: clinical classification to molecular delineation in Sotos syndrome and Beckwith-Wiedemann syndrome. Curr Opin Pediatr. [Internet]. 2005 [citado 28 Mar 2012]; 17(6): [Aprox. 6p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/16282780
26. Mussa A, Chiesa N, Porta F, Baldassarre G, Silengo MC, Ferrero GB. The overlap between Sotos and Beckwith-Wiedemann syndromes. J Pediatr. [Internet]. 2010 [citado 28 Mar 2012]; 156(6): [Aprox. 1p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20394943
27. Weaver Syndrome 1 [Internet]. OMIN: Johns Hopkins University; 2012 [citado 28 Mar 2012]. Disponible en:http://omim.org/entry/277590
28. Basel-Vanagaite L. Acute lymphoblastic leukemia in Weaver syndrome. Am J Med Genet A. [Internet]. Feb 2010 [citado 28 Mar 2012]; 152(2): [aprox. 4 p.]. Disponible en: http://dx.doi.org/10.1002/ajmg.a.33244
29. Ostos Alfonso H, Jara J, Ávila V. Síndrome Weaver: presentación de casos. Iatreia. [Internet]. 2010 [citado 28 Mar 2012]; 23(4). Disponible en: http://medicina.udea.edu.co/ojs/index.php/iatreia/article/viewArticle/1658
30. Rojas Betancourt I. Conferencia sobre Asesoramiento Genético. Ciudad de la Habana: Centro Nacional de Genética Médica; 2005.
Recibido: 11 de junio 2012.
Aprobado: 20 de noviembre 2012

{kind=link}
{kind=link}