










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.17 no.1 Pinar del Río ene.-feb. 2013
ARTÍCULO ORIGINAL
Estrategia de evaluación genética en el Síndrome West
A strategy of genetic assessment in Wests syndrome
Deysi Licourt Otero1, Anitery Travieso Téllez2
1Especialista de Primer Grado en Medicina General Integral, Especialista de Primer y Segundo Grados en Genética Clínica. Centro Provincial de Genética Médica. Asistente. Correo electrónico: deysili@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral y Especialista de Primer Grado en Genética Clínica. Centro Provincial de Genética Médica. Instructora. Correo electrónico: any0511@princesa.pri.sld.cu
RESUMEN
Introducción: La epilepsia ocupa el segundo lugar entre las enfermedades neurológicas de la infancia y produce afectaciones en las esferas afectiva, cognitiva y social de quienes la padecen, así como en su contexto familiar.
Objetivo: diseñar una estrategia para la evaluación genética del Síndrome West.
Material y método: se realizó un estudio descriptivo, transversal en pacientes con diagnóstico de Síndrome West atendidos en el Centro Provincial de Genética Médica de Pinar del Río desde el primero de enero del 2010 al 31 de Agosto del 2011.
Resultados: predominó el Síndrome West en el sexo masculino, con debut de los síntomas entre 4 y 6 meses. Se obtuvo una alta correspondencia entre el diagnóstico de la enfermedad y la identificación de antecedentes prenatales positivos. La amenaza de aborto, el parto pretérmino y la hipoxia neonatal fueron las causas perinatales más atribuidas al desarrollo de la enfermedad. El examen físico dismorfológico fue positivo en la mayoría de los pacientes y aportó elementos que ofrecieron el diagnóstico en casos sin etiología definida. Las pruebas metabólicas y cromosómicas, resultaron útiles en la identificación etiológica del Síndrome West. Se diseñó una estrategia de evaluación genética para los pacientes con Síndrome West.
Conclusiones: la caracterización del Síndrome West según los protocolos de estudio, facilitó el manejo de forma integral, permitió identificar las causas responsables del trastorno y se diseñó la estrategia para la evaluación genética de los niños con esta enfermedad.
DeCS: Síndrome West/etiología/diagnóstico, Epilepsia genética/clasificación/diagnóstico.
ABSTRACT
Introduction: epilepsy occupies the second place among neurological diseases in childhood and it provokes affectations in the emotional, cognitive and social spheres of epilepsy sufferers, as well as in their familial context.
Objective: to design a strategy of genetic assessment in West’s syndrome.
Material and Method: a descriptive and cross-sectional study was conducted in patients suffering from West’s syndrome attended at Provincial Medical Genetics Center in Pinar del Rio from January 1, 2010 - August 31, 2011.
Results: West’s syndrome prevailed in male sex and the onset of symptoms by 4 and 6 months. A high correspondence was found between diagnosis of the disease and the identification of positive prenatal history. Threatened abortion, preterm labor and neonatal hypoxia were the perinatal causes ascribed to the development of the disease. Dysmorphological physical examination was positive in the majority of patients and it provided elements that helped with diagnosis of cases without presenting a definite etiology. Metabolic and chromosomal tests were valuable to perform the etiological identification of West’s syndrome. A strategy to carry out the genetic assessment for West’s syndrome patients was designed.
Conclusions: West’s syndrome characterization following the protocols of study eased a comprehensive management which allowed the identification of causes and the design of a strategy to complete the genetic assessment of children suffering from this disease.
DeCS: Infantile spasm etiology/diagnosis, Epilepsy/classification/diagnosis.
INTRODUCCIÓN
La contribución de la genética en el diagnóstico y seguimiento de las epilepsias y los síndromes epilépticos no se discute en la actualidad. Los servicios de referencia en Neurología a nivel mundial incluyen la evaluación y los estudios genéticos dentro del organigrama habitual para el diagnóstico etiológico y el manejo de los pacientes con epilepsia, tenga esta una causa desconocida o "aparentemente identificada".1
Una de las formas de presentación de la epilepsia de comienzo en la infancia temprana, cuya aparición se relaciona con la edad y que en sí misma no puede considerarse una entidad nosológica es el Síndrome West; constituyendo un heterogéneo grupo de condiciones que comparten la triada clínica de espasmos epilépticos, deterioro del neurodesarrollo y patrón de hipsarritmia en el electroencefalograma (EEG).
La discapacidad cognitiva que se presenta en el 90% de los casos afectados puede ser de diversos grados y con frecuencia se asocia también a déficit motor, a trastornos de conducta y rasgos autísticos. Uno de sus aspectos clínicos más sobresalientes se centra en la magnitud de las consecuencias que produce en el orden del neurodesarrollo este tipo peculiar de síndrome epiléptico.2 El pronóstico, aunque generalmente grave, resulta incierto dada las peculiaridades evolutivas de esta entidad y debido a la multiplicidad de sus bases patológicas.2
El Síndrome de West es una encefalopatía epiléptica considerada como una enfermedad compleja, pudiendo llegar a una discapacidad, lo que conlleva a una enorme repercusión social y económica. No se disponen de estimados seguros de la frecuencia de la enfermedad y se carece de datos sobre las características clínico-genéticas de la enfermedad. En Cuba los estudios sobre el tema son muy escasos e insuficientemente actualizados, por ello la atención genética de la enfermedad es insuficiente para la intervención correcta, efectiva y oportuna en el territorio. El incremento de la calidad de vida en la población, el desarrollo de herramientas para el asesoramiento genético, el desarrollo de las especialidades médicas, el mejoramiento de los servicios de salud y educación, que incluyen la rehabilitación, motivan al continuo esfuerzo por el dominio de la enfermedad y la posibilidad de incorporar a los afectados a las actividades estudiantiles, laborales y sociales.2,3
En la provincia, aún se investiga poco sobre el comportamiento epidemiológico y las bases genéticas de la epilepsia, situación que compromete el diagnóstico etiológico del Síndrome West y por tanto el manejo individualizado y con un enfoque genético de estos pacientes. El objetivo del presente trabajo es diseñar una estrategia para la evaluación genética del Síndrome West mediante la caracterización clínica y epidemiológica.
MATERIAL Y MÉTODO
Se realizó un estudio observacional, descriptivo de corte transversal en una muestra de 33 pacientes con diagnóstico clínico y electroencefalográfico de Síndrome West, atendidos en el Hospital Pediátrico Pepe Portilla y en el Centro Provincial de Genética Médica desde el período de tiempo comprendido entre el primero de enero del 2010 y el 31 de agosto del 2011.
El universo estuvo constituido por el total de pacientes con Síndrome de West que asistieron a los servicios de genética clínica y a las consultas de seguimiento de neurología, estudiándose en su totalidad.
La selección de la muestra se realizó por un muestreo no probabilístico intencionado, teniendo en cuenta criterios de inclusión y exclusión, quedando constituida por 33 niños.
Criterios de Inclusión: Todos los pacientes menores de seis años de edad, que pertenecían a la provincia y que los padres dieron el consentimiento informado.
Criterios de exclusión: Aquellos inasistentes por cualquier causa a las evaluaciones programadas y los pacientes que los padres no dieron el consentimiento informado.
Obtención de la información: Tuvo lugar a partir de tres fuentes principales: revisión documental, entrevista y examen físico e informes de los resultados de estudios realizados. Los pacientes en consulta de Genética Clínica, se les realizó revisión de sus respectivas historias clínicas hospitalarias. Se entrevistó a los familiares, indagando sobre antecedentes familiares de epilepsia y se procedió al examen físico de los casos. Los niños con hallazgos positivos al examen físico dismorfológico, se sometieron a estudios genéticos metabólicos y/o cromosómicos. Se realizó el árbol genealógico de cada paciente como parte de la historia clínica. El estudio citogenético de alta resolución en sangre periférica (cariotipo) se realizó en el Centro Provincial de Genética Médica de Pinar del Río. Para la realización de las pruebas metabólicas en orina se recogió una muestra de la primera micción en ayunas.
Se confeccionó una estrategia para la evaluación genética de los pacientes con Síndrome West. Los resultados se presentaron en tablas y gráficos con la ayuda del procesador de texto Microsoft Word. Los resultados del análisis de las variables cualitativas se resumieron en frecuencias absolutas y por cientos.
RESULTADOS
De los 33 pacientes del estudio, con Síndrome West, 15 son del género femenino para un 45.4% y el 54.5 % corresponden al masculino.
De acuerdo a la edad de comienzo de los primeros síntomas se subdividieron los pacientes en tres grupos de edades. Predominaron los casos cuyo debut de la epilepsia se produjo entre los 4 y los 6 meses de vida, quienes en su totalidad representan más de la mitad de la muestra en estudio. Tabla 1.

Al indagar sobre la historia familiar de epilepsia entre los pacientes con Síndrome West, se encontró que más de la mitad presentaron antecedentes familiares positivos (20/33). Los familiares de primer grado de consanguinidad resultaron ser mayormente los afectados y entre estos existe un mayor porciento por la línea materna (24.2%). Tabla 2
Durante el interrogatorio se exploró cuidadosamente la existencia de eventos prenatales, perinatales o postnatales que pudieran asociarse al debut de los espasmos infantiles. El 54.5% de los casos presentó algún antecedente prenatal positivo, predominando las enfermedades maternas durante el embarazo y la amenaza de aborto y de parto pretérmino (18.1%), seguido por la amenaza de aborto asociada a enfermedad materna (9.0%), el resto de forma individual representó un 3.0%. Dentro de las enfermedades maternas más frecuentes predominó la hipertensión arterial (HTA) crónica o asociada al embarazo que se presentó en 4 de 18 casos con Síndrome West representando el 22.2% y otras enfermedades maternas asociadas a la gestación en 2 de los 18 casos para un 11.1%. Gráfico 1.

Sobre los antecedentes perinatales, el evento más relacionado con el diagnóstico de Síndrome West fue el parto distócico y la hipoxia neonatal (41.6 y 33.3 respectivamente). Gráfico 2

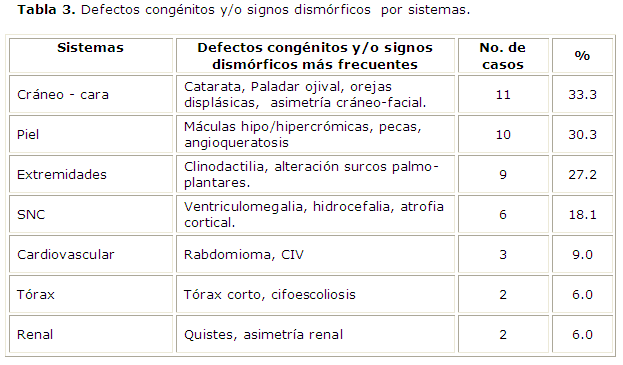
En la tabla 3 se reflejan los defectos congénitos relacionados por sistemas y las características clínicas o signos dismórficos que se encuentran en estos pacientes, de los 33 pacientes del estudio 31 mostraron un examen físico-genético positivo para un 93.9%, resultando que las tres regiones corporales donde se agrupan la mayoría de las dismorfias son el cráneo y la cara, la piel y las extremidades (33.3%, 30.3%, 27,2% respectivamente), algunos pacientes presentan defectos congénitos y/o signos dismórficos de más de un sistema.
Según la presencia o no de defectos congénitos mayores y menores, predominaron los pacientes con defectos menores (75.7%), 14 pacientes mostraron 3 o más defectos menores, mientras que 11 presentaron entre 1 y 2 defectos menores. Los pacientes con defectos mayores resultaron 13 y en todos estos se presentan tres o más defectos congénitos menores. Se comprobó como la presencia de más de tres defectos congénitos menores se asocia generalmente con defectos congénitos mayores. Los síndromes neurocutáneos que se diagnosticaron clínicamente son: la Neurofibromatosis tipo 1 que en la muestra se incluyen siete pacientes, el Complejo Esclerosis Tuberosa que en este estudio se reportan 9 niños, y un niño con Síndrome de Sturge Weber.
Con respecto a los resultados de los estudios cromosómicos realizados, del total de 33 pacientes solo a 25 se les realizó cariotipo de alta resolución, se representa tres casos con variantes cromósomicas normales para un 12 %, dos casos con trisomía para el cromosoma 21 y un caso con una alteración cromosómica estructural del tipo translocación (4 %).
Se realizó estudio metabólico a un total de 17 pacientes. En todos se presentó alteración de las pruebas cualitativas, de estos pacientes solo en un 88.2% se diagnosticó defectos a nivel de los aminoácidos, fundamentalmente la leucina/isoleucina, histidina, glicina y fenilalanina y por último en 2 casos, logró caracterizarse el defecto dado por un niño con fenilcetonuria clásica y una niña con enfermedad de la orina con olor a jarabe de Arce y en el resto quedó planteado el diagnóstico de aminoacidopatía inespecífica.
DISCUSIÓN
Con respecto a la relación de varones y hembras, los datos coinciden con los reportes de las investigaciones relacionadas sobre el tema que llegan a tener indicadores de hasta 3 varones por cada hembra. 3,4 Se ha expuesto que el cromosoma X contiene numerosos loci relacionados potencialmente con el desarrollo del Sistema Nervioso Central. Así mismo se reporta la mayor frecuencia de varones afectados con discapacidad intelectual, trastornos de conducta o retrasos específicos en determinadas esferas del conocimiento como es el caso del lenguaje. 5
Algunos autores reportan en su estudio la relación entre la edad de debut de los primeros síntomas del Síndrome West y las causas de este. Sugieren que en dependencia de la etiología identificada para el síndrome epiléptico será más temprano o no el inicio de los síntomas, dado el momento del desarrollo del SNC en que se produjo el daño. 6
Según la distribución de pacientes con antecedentes familiares positivos de epilepsia por vía materna y paterna el Síndrome West se ha relacionado con mecanismos no mendelianos y es necesario remarcar el concepto conocido como "imprinting" genómico, que puede dar lugar a un fenotipo diferente según la anomalía proceda del padre o de la madre. 7,8
Numerosos reportes se han publicado referidos a la asociación entre exposición a factores intrauterinos y desarrollo de enfermedades postnatales, dentro de las que se incluye la epilepsia. 9,10 Los antecedentes perinatales positivos se comportan como en otros muchos estudios que relacionan los eventos alrededor del nacimiento con el debut de Espasmo infantiles. 11 Concretamente estos autores alertan hacia la minuciosa evaluación de pacientes con eventos perinatales, pues puede ocurrir que muchos de ellos no sean tal como se califican sino que tengan un antecedente prenatal más frecuente de lo que habitualmente se sospecha.
Con respecto a los defectos congénitos y/o signos dismórficos por sistemas, durante la exploración física de los pacientes incluidos en la muestra se constató el valor insustituible del examen físico minucioso en los trastornos del neurodesarrollo, pues los hallazgos sugieren las posibles causas subyacentes, sobre todo las de origen genético de comienzo prenatal que dejan su huella física de acuerdo al momento del desarrollo en que produjeron las alteraciones. 9,10
Se comprobó como la presencia de más de tres defectos congénitos menores se asocia generalmente con defectos congénitos mayores. Los síndromes neurocutáneos diagnosticados clínicamente como la Neurofibromatosis tipo 1 que en la muestra se incluyen siete pacientes, y la Esclerosis tuberosa que en este estudio se reportan 9 niños, así como un niño con Síndrome de Sturge Weber, se reportan entre las asociaciones etiológicas del Síndrome West. 4,5 Los estudios más recientes en Estados Unidos y el Reino Unido donde se asegura que con los datos de la historia individual, el examen físico y los estudios de imagen se logra identificar la etiología de aproximadamente el 70% de los casos con Síndrome West. 12.
Los resultados obtenidos en los estudios cromosómicos, apoyan la propuesta del Grupo de Trabajo para los Espasmos infantiles (ISWG), que sugiere la realización de estudio cromosómico a todo niño con Espasmos infantiles y dismorfias, que luego de realizada la historia individual, el examen físico y las exploraciones imagenológicas se mantenga sin causa definida para su trastorno. 12
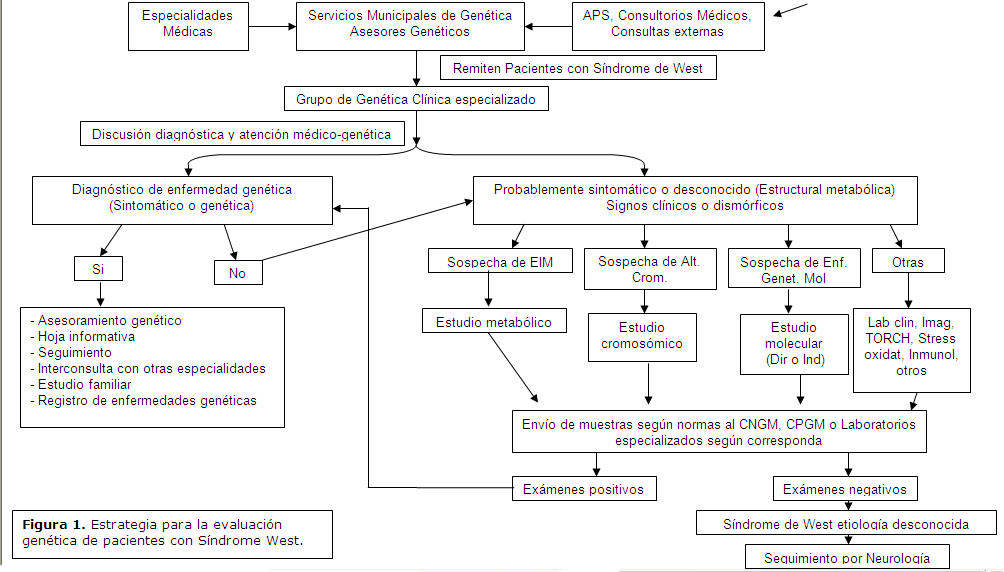
Cuando cada paciente se maneja de forma integral y según los protocolos de estudio, las causas responsables del trastorno se definen progresivamente. Por ello se propone la siguiente estrategia para la evaluación Genética de los pacientes con Síndrome West. (Figura 1).
REFERENCIAS BIBLIOGRÁFICAS
1. Pascual GJ, Oliver LS, Marrero MP, García GR, Borbolla VL, García MD. Editores. Pediatría. Vol. 4. Ciudad de la Habana: Editorial Pueblo y Educación; 1999.
2. Gurnet CA, Hedera P. New ideas in epilepsy genetics: novel epilepsy genes, copy number alterations, and gene regulation. Arch Neurol 2007; 64(3): 324-8.
3. Lúthvígsson P, Olafsson E, Sigurthardóttir S, Hauser WA. Epidemiologic features of infantile spasms in Iceland. Epilepsia[en línea]. 1994[fecha de acceso 21 de noviembre de 2010]; 35(4):802-5. Disponible en: http://hinari-gw.who.int/whalecomonlinelibrary.wiley.com/
4. Hino-Fukuyo N, Haginoya K, Iinuma K. Epidemiological and clinical studies of West syndrome in Miyagi Prefecture, Japan. No To Hattatsu[en línea]. 2007[fecha de acceso 13 de marzo de 2010]; 39(4):257-61. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed
5. Laumonnier F, Cuthbert PC, Seth GN. The Role of Neuronal Complexes in Human X-Linked Brain Diseases. Am J Hum Genet. [en línea]. 2007 February[fecha de acceso 12 de junio de 2010]); 80(2): 205-220. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed
6. Hamano S, Tanaka M, Mochizuki M. Long term follow up study of West syndrome: differences of outcome among symptomatic etiologies. J Pediatr[en línea]. 2003[fecha de acceso 29 de diciembre de 2009]; 143:231-235. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed
7. Sisodiva S, Cross JH, Blumcke I. Genetics of epilepsy: Epilepsy Research Foundation workshop report. Epileptic Disord 2007; 9(2): 194-236.
8. Hansen CP, Moller R, Tumer Z. The genetic basis of epilepsy. The Danish Epilepsy Society. Ugeskr Laeger 2007; 169(12): 1102.
9. Sarit Avishai-Eliner, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci[en línea]. 2002[fecha de acceso 3 de abril de 2010]; 25(10): 518-524. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed
10. Cottrell EC, Seckl JR. Prenatal Stress, Glucocorticoids and the Programming of Adult Disease. Behav Neurosci. 2009; 3: 19
11. González AE, Díaz A, Díaz-Anzaldúa A. La epigenética y los estudios en gemelos en el campo de la psiquiatría. Salud Mental 2008; 31:229-37.
12. Osborne JP, Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, et al. The underlying etiology of infantile spasms (West syndrome): Information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia[en línea]. 2010 [fecha de acceso 1 de diciembre de 2010]; 51(10):2168-2174. Disponible en: http://hinari-gw.who.int/whalecomonlinelibrary.wiley.com.
Recibido: 13 de noviembre 2012.
Aprobado: 21 de enero 2013.
Dra. Deysi Licourt Otero. Especialista de Primer Grado en Medicina General Integral, Especialista de Primer y Segundo Grados en Genética Clínica. Centro provincial de Genética Médica. Asistente. Correo electrónico: deysili@princesa.pri.sld.cu. KM 89 Carretera Central. Pinar del Río. Cuba.

{kind=link}
{kind=link}
{kind=link}