










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versión On-line ISSN 1561-3194
Rev Ciencias Médicas vol.22 no.2 Pinar del Río mar.-abr. 2018
ARTÍCULO ORIGINAL
Comportamiento clínico y complicaciones de la hemofilia en la población pediátrica
Clinical behavior and complications of hemophilia in the pediatric population
Jorge Luis Hernández Gonzalez,1 Mirta Campo Díaz,2 Cesar Valdés Sojo,3 Grettel Borrego Cordero,4 Claudia Cabrera Morales 5
1 Médico Especialista de Primer Grado en Hematología. Máster en Atención Integral al Niño. Profesor asistente. Hospital Pediátrico Provincial Pepe Portilla. Pinar del Río. Cuba. jorgel@infomed.sld.cu
2 Médica. Especialista de Segundo Grado en Hematología y en Pediatría. Máster en Atención Integral al Niño. Profesora Auxiliar. Investigadora
auxiliar. Profesora Consultante. Hospital Pediátrico Provincial Pepe Portilla. Pinar del Río. Cuba. jorgel@infomed.sld.cu
3 Médico. Especialista de Primer Grado en Medicina General Integral y en Hematología. Máster en Atención Integral al Niño con Enfermedades Hematológicas Crónicas. Instructor. Hospital Pediátrico Provincial Pepe Portilla. Pinar del Río. Cuba. cvsojo@infomed.sld.cu
4 Médica. Especialista de Primer Grado en Medicina General Integral y en Hematología. Máster en Atención Integral al Niño con Enfermedades Hematológicas Crónicas. Hospital Pediátrico Provincial Pepe Portilla. Pinar del Río. Cuba. jorgel@infomed.sld.cu
5 Médica. Residente de primer año en Hematología. Instituto de Hematología e Inmunología. La Habana. Cuba. lau.93@nauta.cu
Recibido: 07 de octubre de 2017
Aprobado: 09 de enero de 2018
RESUMEN
Introducción: la hemofilia es una enfermedad hereditaria ligada al sexo, producida por disminución de la actividad coagulante del factor VIII (hemofilia A), del factor IX (hemofilia B) y del factor XI (hemofilia C). Su tratamiento representa un elevado costo económico para el país y se asocia con gran frecuencia a discapacidades serias del paciente afecto, impactando negativamente en su desarrollo biopsicosocial, así como en la adecuada funcionabilidad familiar.
Objetivo: caracterizar clínica y epidemiológicamente la hemofilia en la población pediátrica de la provincia Pinar del Río, en el periodo de enero 1985 a junio de 2016.
Método: se realizó un estudio observacional, descriptivo y transversal en pacientes pediátricos diagnosticados con hemofilia. Los datos fueron extraídos de los expedientes clínicos. El diagnóstico se estableció mediante coagulograma, dosificación de factores plasmáticos VIII, IX y XI y dosificación de inhibidores. Se empleó estadística descriptiva.
Resultados: de un total de nueve individuos hemofílicos se observó un franco predominio de la A (n=8) y de la forma moderada (n=5). Los diagnósticos se realizaron antes de un año de edad (66%), donde las hemorragias traumáticas (sangrados intracraneales) fueron las más frecuentes (22.1%). Todos los pacientes con comportamiento moderado o severo tuvieron sangrado músculo-esquelético. Las principales complicaciones de la enfermedad fueron epilepsia secundaria y retraso mental como secuela de hemorragia intracraneal neonatal. Las complicaciones terapéuticas estuvieron relacionadas con procesos aloinmunes.
Conclusiones: las hemorragias traumáticas en etapas tempranas de la vida fueron la causa más frecuente del diagnóstico de la enfermedad, siendo el sangramiento del sistema nervioso central la principal causa de secuelas en estos pacientes.
DeCS: HEMOFILIA A; NIÑO; EPIDEMIOLOGÍA.
ABSTRACT
Introduction: hemophilia is an inherited disease linked to sex, caused by the decreased coagulant activity of factor VIII (hemophilia A), factor IX (hemophilia B) and factor XI (hemophilia C). Its treatment represents a high economic cost for the country and it is associated with great frequency to serious disabilities of the afflicted patient, negatively impacting its bio-psychosocial development, as well as an adequate familiar functionality.
Objective: to characterize hemophilia clinically and epidemiologically in the pediatric population of Pinar del Río province, in the period from January 1985 to June 2016.
Method: an observational, descriptive and cross-sectional study was conducted in pediatric patients diagnosed with hemophilia. The data were extracted from the clinical files. The diagnosis was established by coagulogram, dosage of plasma factors VIII, IX and XI and dosage of inhibitors. Descriptive statistics was used.
Results: out of a total of 9 hemophiliac individuals, a clear predominance of A (n = 8) and moderate form (n = 5) was observed. The diagnoses were made before one year of age (66 %), where traumatic hemorrhages (intracranial bleedings) were the most frequent (22.1%). All patients with moderate or severe behavior had muscle-skeletal bleeding. The main complications of the disease were secondary epilepsy and mental retardation as a consequence of neonatal intracranial hemorrhage; therapeutic complications were related to alloimmune processes.
Conclusions: traumatic hemorrhages in the early stages of life were the most frequent cause of the diagnosis of the disease, with bleeding from the central nervous system being the main cause of sequelae in these patients.
DeCS: HEMOPHILIA A; CHILD; EPIDEMIOLOGY.
INTRODUCCIÓN
La hemofilia es una enfermedad hereditaria ligada al sexo producida por la disminución de la actividad del factor VIII (hemofilia A) o del factor IX (hemofilia B). Ambos genes se encuentran en el cromosoma X: el gen del factor VIII (FVIII) al final del brazo largo, en la región Xq28 y el gen del factor IX (FIX); también en el brazo largo pero más cercano al centrómero, en la región Xq27.(1) También se describen casos con disminución de la actividad del factor XI (hemofilia C) que se diferencia de las anteriores por heredarse de forma autosómica recesiva, con igual frecuencia en ambos sexos.(2)
Las hemofilias A y la B son clínicamente semejantes, ya que en ambas se altera el paso esencial de la generación de trombina y la severidad de las manifestaciones hemorrágicas mantiene una relación directa con la actividad del factor en el plasma. Solo los pacientes graves sufren sangrados espontáneos, siendo necesario enfatizar que aun los enfermos leves sangran abundantemente después de traumatismos o cualquier procedimiento invasivo. Las hemartrosis son las más características de esta entidad y las más invalidantes.(1-3)
El diagnóstico de la hemofilia se establece por los antecedentes familiares y personales, las manifestaciones clínicas y el estudio de laboratorio. Se sospecha por la presencia de un tiempo parcial de tromboplastina activado (TPT), prolongado y se confirma con la disminución de la actividad coagulante del factor VIII (FVIII), factor IX (FIX) o factor XI (FXI). En la actualidad es posible realizar el diagnóstico prenatal y de portadora de hemofilia por técnicas de análisis del ADN en las embarazadas de riesgo.(2-4)
La incidencia de hemofilia A es de 1 en 10000 varones y la de hemofilia B es de 1 en 60000.(1) Hasta el año 2013 y con un 91% representatividad de las naciones esta entidad nosológica había sido diagnosticada a 176 211 personas, de los cuales 415 eran cubanos, para una tasa incidental nacional de 0.004 %.(5)
Se encuentra incluida dentro del grupo de las enfermedades raras, pero su tratamiento representa un elevado costo económico para el país y se asocia con gran frecuencia a discapacidades serias del paciente afecto, impactando negativamente en su desarrollo biopsicosocial, así como en la adecuada funcionabilidad familiar. Estos motivos justifican la importancia del estudio, la introducción de los resultados evidenciará una mejora en la atención médica integral que se brinda en la actualidad al trastorno congénito de la coagulación que con mayor frecuencia se diagnostica.
La caracterización clínico-epidemiológica y de los estudios de la coagulación de los niños con hemofilias tiene entre sus objetivos la correcta tipificación y clasificación,(3) que unido a la atención integral de estos pacientes, la detección temprana de sus complicaciones y las correcciones de las mismas junto al apoyo psicológico, familiar y escolar constituyen pilares fundamentales para el desarrollo biopsicosocial de los niños.
El propósito de este trabajo ha sidocaracterizar clínica y epidemiológicamente la hemofilia en la población pediátrica de la provincia Pinar del Río, en el período desde enero de 1985 a junio de 2016.
MÉTODO
Se realizó un estudio observacional, descriptivo y transversal para caracterizar clínica y epidemiológicamente la hemofilia en la población pediátrica de la provincia de Pinar del Río, Cuba, durante el período enero de 1985 - junio de 2016. Se incluyeron todos los pacientes (N=9) que fueron diagnosticados en el periodo descrito y tratados en consulta de Hematología del Hospital Pediátrico Provincial Docente "Pepe Portilla" de Pinar del Río, Cuba.
Las variables que se estudiaron fueron: tipo de hemofilia (A por deficiencia del factor VIII de la coagulación, B por deficiencia del factor IX o C por deficiencia del factor XI), considerando como valores normales de la dosificación el intervalo 0.6 a 1.2 uac/mlPF; severidad de la hemofilia (leve cuando la actividad del factor deficiente pertenece al intervalo 6-30 %, moderada sí está entre 1-5 % o severa cuando es inferior a 1%); forma clínica de presentación (hemorragia intracraneal, hematoma traumático, hemartrosis, hematoma del cargado, brote dentario o sangrado traumático); momento de debut (periodo neonatal, lactante o mayor de 1 año); manifestaciones clínicas en el curso de la enfermedad; complicaciones relacionadas con la enfermedad y complicaciones relacionadas con el tratamiento.
La metodología diagnóstica estuvo sustentada en la realización de un coagulograma utilizando un coagulómetro de cuatro canales marca Start de la Diagnóstica Stago. A los pacientes con prolongación del tiempo parcial de tromboplastina se les realizó dosificación de factores plasmáticos VIII, IX y XI para confirmar el tipo de hemofilia y su severidad. Se realizó además dosificación de inhibidores para clasificarlos en alto o bajo respondedor.
La información se obtuvo de los expedientes clínicos de los enfermos. Los datos obtenidos fueron resumidos y procesados en una base de datos automatizada con campos creados para cada una de las variables y el Epidat versión 3.1 para resumir la información en medidas de frecuencias absolutas y relativas.
Para la realización de la investigación se consultó el Comité de Ética de la institución que mostró su aprobación. Se cumplieron los principios éticos de autonomía, beneficencia y justicia mediante la obtención de su consentimiento escrito. Se declaró que la investigación no revelaría la identidad de los pacientes. Los datos y resultados solo serán expuestos en marcos de interés científico.
RESULTADOS
Del total de 9 pacientes diagnosticados se encontró un franco predominio de la hemofilia A (8/9; 88.9 %) y de la forma moderada de la enfermedad presente en 5 pacientes (tabla 1). No se detectaron pacientes con hemofilia C.
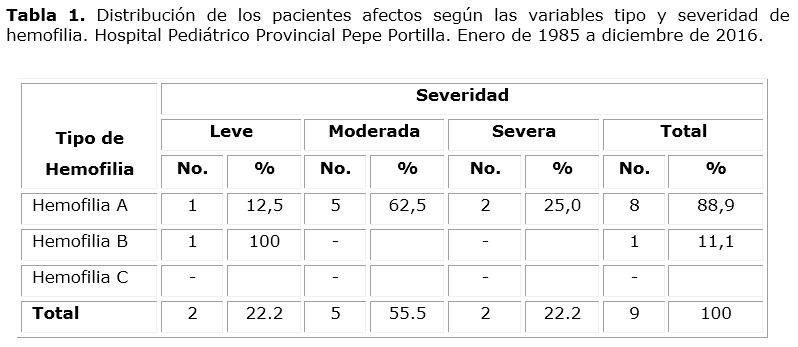
Resulta importante resaltar que no se encontraron antecedentes de la enfermedad en el 33.3 % de los pacientes diagnosticados. En estos casos a los hombres de la línea materna se les realizó dosificación de factores, con resultados dentro de límites normales, lo que permitió excluir la posibilidad de la existencia de la deficiencia en las familias; los enfermos corresponden 2 a hemofilia A y 1 a hemofilia B.
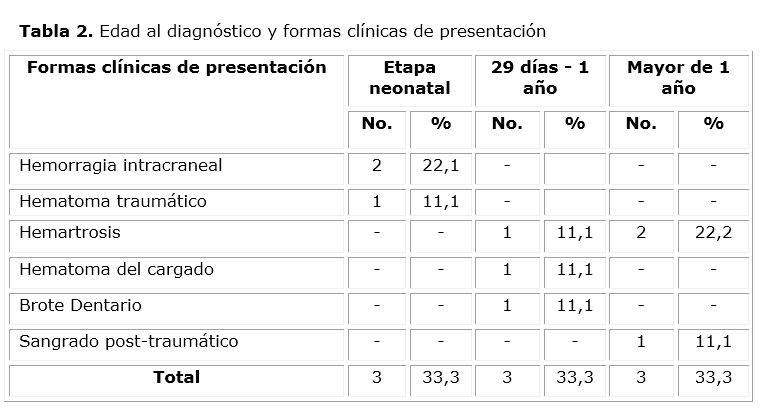
El diagnóstico se realizó antes del primer año de edad, con igual número de casos en el periodo neonatal y en el de lactante en número de 3; encontrándose diferencias en la forma clínica de debut, siendo mayor la hemorragia intracraneal en el primer momento y las hemartrosis, hematoma del cargado y el brote dentario en el segundo momento. Posterior a esta edad, la principal causa del diagnóstico fue la hemartrosis relacionada con el inicio de la deambulación (tabla 2).
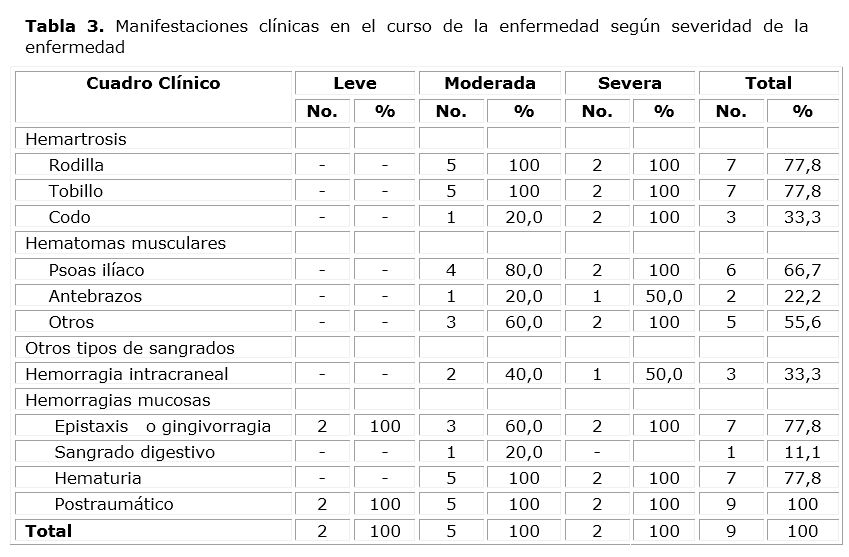
Todos los pacientes con independencia de la severidad de la enfermedad han presentado al menos un episodio de sangrado luego de su diagnóstico. Los pacientes con severidad moderada y grave han sido los más afectados con respecto a las recidivas de la enfermedad; las hemartrosis y los hematomas musculares son las manifestaciones clínicas que con mayor frecuencia han sido encontradas en las reconsultas (tabla 3).
Se encontraron complicaciones relacionadas con la enfermedad en solo dos pacientes con epilepsia secundaria y retraso mental como secuela de hemorragia intracraneal neonatal e igual cantidad con artropatías deformantes.
Las únicas complicaciones relacionadas con el tratamiento estuvieron relacionadas con procesos a los inmunes como la adquisición de inhibidores (3/9; 33,3 %) y el shock anafiláctico por el uso de crioprecipitado (1/9; 11,1 %). En ningún paciente se detectaron enfermedades virales trasmitidas por sangre.
DISCUSIÓN
La hemofilia es la coagulopatía congénita más representada a escala global.(5) Su incidencia es de aproximadamente 1:5 000 nacimientos masculinos y su prevalencia de 10 a 20 casos por cada 100 000 varones, de los cuales el 80 % padecen hemofilia A y un 20 % hemofilia B. (1)Alrededor del 25 % de los pacientes hemofílicos no refieren antecedentes familiares de la enfermedad, lo que habitualmente está asociado a mutaciones genéticas "de novo".(4,5) Estos reportes se corresponden con los resultados encontrados en la serie estudiada.
La hemofilia se clasifica en leve, moderada o severa de acuerdo al nivel residual de la actividad plasmática del factor deficiente. El estudio encontró que el 55 % de los afectos se correspondían con hemofílicos moderados, los cuales presentan sangrados con elevada frecuencia. Esto se corresponde con lo expuesto por otros autores, (3,6,7) que estiman que la mediana en el número de episodios hemorrágicos por año es de uno para los pacientes moderados y de 6 para los pacientes con hemofilia severa, así como sangrados prolongados secundarios a traumatismos o cirugía, en aproximadamente el 70 % de los pacientes.
Es destacable que aunque los sangrados espontáneos son muchos más frecuentes en los hemofílicos severos, el comportamiento en la provincia de los pacientes hemofílicos moderados con una dosificación de dos a tres porciento mostró un cuadro clínico similar a los catalogados como severos, por lo que consideramos que el porcentaje del factor no debe ser el criterio único para la clasificación del paciente, sino que debe tenerse en cuenta la expresión clínica individual de la enfermedad, sus complicaciones y las secuelas que pudieran contribuir al resangrado en determinados sitios.
En la actualidad es posible llevar a cabo el diagnóstico prenatal de la enfermedad mediante la realización de estudios genéticos en familias de riesgo, aunque existen numerosos factores que lo dificultan.(4,6) Todos los casos estudiados fueron diagnosticados después del nacimiento, de ellos seis antes del primer año de vida.
Los traumas obstétricos y los procedimientos invasivos cuando se desconocen los antecedentes patológicos familiares de hemofilia pueden causar hemorragias desproporcionadas que conducen a la realización de estudios de la coagulación, y es entonces cuando se realiza el diagnóstico. Sin un tratamiento rápido y efectivo las hemorragias se traducen en discapacidades que impactan seriamente la calidad de vida. (8)
El sangrado por el cordón umbilical es referido como el primer sitio de sangramiento en los hemofílicos severos, pudiéndose presentar luego hemorragias intracraneales. El sangrado articular o muscular habitualmente ocurre cuando el niño inicia la deambulación, (7) encontrándose semejanzas con los hallazgos del estudio.
En las etapas posteriores de la vida los sangrados musculares y articulares pasan a ocupar las manifestaciones clínicas más frecuentes en la población hemofílica, los mismos pueden ser muy dolorosos, debilitantes, y cuando se repiten en un mismo sitio, pueden presentar complicaciones a largo plazo, tales como sinovitis crónica, artropatía deformante, contracturas, formación de pseudotumores (en el tejido blando y hueso) y fracturas.(3, 8)
König en 1892 fue el primer autor que caracterizó la artropatía hemofílica, separándola del resto de las artritis. Clínicamente el dolor agudo es el síntoma principal producido por la distensión capsular. La radiografía simple mostrará aumento de la densidad de las partes blandas periarticulares, los estudios de ultrasonido y resonancia magnética son de utilidad en caso de duda, principalmente en articulaciones como la cadera y hombro.(8) La principal manifestación clínica en los pacientes estudiados fue la hemartrosis, predominando la toma articular de rodilla y tobillos. Dos de estos pacientes pertenecen al grupo de hemofilia severa con artropatía deformante y anquilosante, y uno de ellos ha desarrollado una deformidad en flexión plantar (equino) que le imposibilita la marcha, estando confinado a un sillón de ruedas. La no disponibilidad en el país de concentrado para el tratamiento quirúrgico corrector y el desarrollo de inhibidores en dicho paciente que además presenta una epilepsia secundaria intratable, secuela de una hemorragia intracraneal traumática, ha impedido hasta el momento actual establecer la terapéutica correctora para dicha complicación.
El hematoma del psoas ilíaco es una afección frecuente, siendo común la compresión del nervio crural con paresia o parálisis del músculo cuádriceps, hipo - o arreflexia rotuliana e hipo - o anestesia en la parte anterior del muslo, que puede extenderse hacia la parte interna de la pierna. Es un hematoma que recidiva con frecuencia y requiere siempre hospitalización con indicación de reposo absoluto en cama en posición de flexión de la cadera.(3)
La incidencia de hematoma del psoas se observó en 6 pacientes de los evaluados en el estudio. Cuatro de ellos han sido incompletos con recuperación total en un periodo que ha oscilado alrededor de un mes. Se evidenció el resangrado en tres de ellos, es de señalar que dos pacientes han tenido un hematoma del psoas completo con evidencia de compromiso neurológico que ha necesitado de tres a cuatro meses para su completa recuperación.
La hemorragia intracraneal es la principal causa de muerte en pacientes hemofílicos. Por fortuna, solo se presenta entre 2-12 % de los enfermos. Se manifiesta de inmediato o 24-48 horas después del trauma. Los datos iniciales incluyen: cefalea, náuseas, vómitos, irritabilidad, crisis convulsivas y signos de focalización neurológica, posteriormente puede aparecer la toma del estado de conciencia. Por esta razón todo traumatismo craneal en un paciente hemofílico debe ser considerado grave y manejarse con reposición temprana, elevando el nivel del factor deficiente a 100 % de actividad, con lo que se previene la hemorragia que constituye un tercio del total de las muertes.(8)
Durante las últimas dos décadas la transmisión del VIH y de la hepatitis viral a través de productos sanguíneos se ha convertido en causa frecuente de morbilidad y mortalidad en la población de pacientes con hemofilia. En la actualidad los productos sanguíneos son atenuados viralmente, pero la adquisición de enfermedades virales por vía sanguínea sigue siendo un importante motivo de preocupación en términos de seguridad. Los factores recombinantes de la coagulación son un paso importante en la seguridad del tratamiento.(3)
Es de señalar que de los nueve pacientes hemofílicos incluidos en la investigación, a pesar de ser politransfundidos con diferentes hemoderivados, ninguno es portador de VIH, virus de la hepatitis B o C, virus que se pesquisan en el país. La vacunación para el virus de la hepatitis B ha sido efectiva y explica la ausencia de esta complicación en la población pediátrica. Esto guarda relación con la aplicación del Programa Nacional de Certificación de la Sangre de Cuba que garantiza el control de toda la sangre donada, lo que disminuye la posibilidad de contagios. (9)
Otra complicación relacionada con la terapéutica es la adquisición de inhibidores del FVIII o FIX que se elevan en etapas tempranas del tratamiento después de nueve a 12 exposiciones al FVIII o FIX con una incidencia aproximada de 15 al 30 % en los pacientes con hemofilia A severa, y en aproximadamente un 5 % de los pacientes con hemofilia B. (10)La severidad de la enfermedad, los antecedentes étnicos del individuo, la historia familiar de inhibidores, el genotipo del antígeno leucocitario humano (HLA) y la mutación que causa la enfermedad están asociados con su desarrollo; también se describen factores ambientales como la edad al momento de la primera exposición al reemplazo de factor que podrían desempeñar algún papel. (11)
En los pacientes hemofílicos evaluados, tres de ellos (dos severos y uno moderado), han desarrollado inhibidores, catalogados los tres pacientes como altos respondedores. Otro de los enfermos presentó un shock anafiláctico durante la administración de crioprecipitado como expresión de una respuesta inmune patológica a dicho hemoderivado. En los pacientes con inhibidores analizados se recogió historia familiar de inhibidores en dos de ellos que han estado expuestos a tratamientos sustitutivos desde etapa neonatal. Consideramos que en los pacientes analizados la aparición de inhibidores ha estado relacionada con el empleo de altas y frecuentes dosis de hemoderivados durante el curso de las terapéuticas establecidas para los sangrados graves o mantenidos que han puesto en peligro la vida del enfermo. El manejo terapéutico actual de los pacientes con altos títulos de inhibidores necesita el uso de concentrado de FVII (Novo Seven®) para el tratamiento de sus episodios hemorrágicos agudos. Su uso también está indicado en la aplicación de protocolos de inmunotolerancia, pudiendo elevar la tasa de éxito y reducir la duración y el costo de la misma en los países donde estos tratamientos puedan ser realizados. (5, 11, 12)
La hemofilia es un padeciemiento crónico que impone alto impacto económico y social a pesar de su baja prevalencia. Los pacientes pediátricos que han sido atendidos en Pinar del Río se caracterizan por una baja incidencia de complicaciones y secuelas, lo que está relacionado con un eficiente trabajo multidisciplinario enmarcado en el protocolo internacional Programa cubano para el diagnóstico y la atención integral de los pacientes con hemofilia cuyo centro rector es el Hospital Pediátrico Provincial Docente Pepe Portilla.
REFERENCIAS BIBLIOGRÁFICAS
1. Castillo González D. Hemofilia II. Aspectos moleculares y de genética poblacional. Rev Cubana Hematol Inmunol Hemoter. [Internet]. 2012 Jun [citado 2017 Sep 14]; 28(2): 111-119.]. Disponible en: http://scielo.sld.cu/pdf/hih/v28n2/hih02212.pdf.
2. MedlinePlus. Hemofilia.2/13/2015 [ Internet]. Disponible en https://www.nlm.nih.gov/medlineplus/spanish/ency/article/000537.htm
3. Rodriguez Merchan EC. .Prevention of the Musculoskeletal Complications of Hemophilia.Advances in Preventive Medicine.Volume 2012 (2012), Article ID 201271. Disponible: http://dx.doi.org/10.1155/2012/201271http://www.hindawi.com/journals/apm/2012/201271/
4. Lavaut Sánchez K. Importancia del diagnóstico de portadoras en familias con antecedentes de hemofilia.. Revista Cubana de Hematología, Inmunología y Hemoterapia [ revista en Internet]. 2014 [citado 2017 Oct 6];30(2):[aprox. 0 p.]. Disponible en: http://www.revhematologia.sld.cu/index.php/hih/article/view/134
5. World Federation of Hemophilia Report on the Annual Global Survey 2013 [ Intrnet] Nov 2014. Disponible en: http://www1.wfh.org/publications/files/pdf-1591.pdf
6. Almagro D, Castillo D, Agramonte O. Guía terapéutica de la hemofilia. Instituto de Hematología e Inmunología. Ciudad Habana: s/n; 2015.p.1-14. Fundación Novonordisk para la hemofilia, Suiza.
7. Castillo González D, Lardoeyt Ferrer R, Almagro Vázquez D, Lam Díaz R, Lavaut Sánchez K, et al. Prevalence of hemophilia in six cuban provinces. Revista Cubana de Hematología, Inmunología y Hemoterapia [ revista en Internet]. 2014 [citado 2017 Oct 6];30(2):[aprox. 0 p.]. Disponible en: http://www.revhematologia.sld.cu/index.php/hih/article/view/145
8. Beeton K. Comment on: "Clinical and functional evaluation of the joint status of hemophiliac adults at a Brazilian blood center".Rev Bras HematolHemoter[ Internet]. 2013 [citado 2016 Oct 15]; 35(1): [aprox. 6 p.].Disponible en: http://www.scielo.br/pdf/rbhh/v35n1/v35n1a03.pdf.
9. Sánchez Frenes P, Fariñas Reinoso AT, Rojo Pérez N, Hernández Malpica S. Diseño de un sistema de vigilancia para infecciones transmitidas por transfusión de sangre en Cienfuegos. Rev Cubana Salud Pública [ revista en Internet]. 2011 [citado 8 Dic 2016];37(2):[aprox. 20p]. Disponible en: http://www.bvs.sld.cu/revistas/spu/vol37_02_11/spu13211.htm
10. Halimeh S, Bidlingmaier Ch, Heller Ch, Gutsche S, Holzhauer S, Kenet G,et al. Clinical Study Risk Factors for High-Titer Inhibitor Development in Children with Hemophilia A: Results of a Cohort Study. BioMed Research International [ Internet]. 2013. Disponible en: http://www.hindawi.com/journals/bmri/2013/901975/.
11. Salinas Escudero G, Galindo-Suárez RM, Rely K, Carrillo Vega MF, Muciño E. Análisis del costo y la efectividad de los esquemas de administración de factores de coagulación para el manejo de niños con hemofilia A en México. Bol Med Hosp Infant Mex [ Internet]. 2013 Jul–Ago [citado 2013 Oct 14]; 70(4): [aprox. 11 p.]. Disponible http://www.google.com.cu/url?q=http://www.medigraphic.com/pdfs/bmhim/hi-2013/hi134d.pdf&sa=U&ei=shwOU7SVLrPKsQTtmoDYBQ&ved=0CB8QFjAA&usg=AFQjCNFrYLjdYTKPvK9epv15nmu6fW96zw
12. Castillo González D, Machín García S, Macías Pérez I, Agramonte LLanes OM, González-Otero A, Serrano-Mirabal J. Hemorragias poco usuales en pacientes con hemofilia. Revista Cubana de Hematología, Inmunología y Hemoterapia [ revista en Internet]. 2015 [citado 2017 Oct 6];31(2):[aprox. 0 p.]. Disponible en: http://www.revhematologia.sld.cu/index.php/hih/article/view/253
Jorge Luis Hernández Gonzalez: Médico Especialista de Primer Grado en Hematología. Máster en Atención Integral al Niño. Profesor asistente. Hospital Pediátrico Provincial Pepe Portilla. Pinar del Río. Cuba. Si usted desea contactar con el autor de la investigación hágalo aquí

