










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome Klippel y Trénaunay (SKT) constituye una rara y compleja malformación vascular congénita, de flujo lento, que se caracteriza por la triada clínica de nevos, hipertrofia ósea y de partes blandas de la zona corporal involucrada y varicosis atípica de una extremidad o venas periféricas laterales anómalas e hipertróficas.1) Según las recomendaciones de la Sociedad Internacional para el estudio de Anomalías Vasculares, se mantiene la clasificación de este síndrome dentro del grupo de las anomalías vasculares combinadas2 y tiene una baja incidencia del orden de 1:100 000 recién nacidos.3
La etiología no se conoce del todo, por lo mismo constituye una entidad esporádica. Se ha postulado que es el resultado de un trastorno de desarrollo embrionario de los tejidos mesodérmicos que afectan la angiogénesis en diferentes etapas, muy probable después de una lesión intrauterina.4
Los pacientes muestran un cariotipo normal, sin embargo, se informan traslocaciones en los cromosomas 5 al 11 y 8 al 14, así como el cromosoma 18 supernumerario que sugieren la posibilidad de mutación única.5 También se asocia a una mutación en gen PIK3CA, involucrado en la angiogénesis y con mosaicismo somático. Se plantea un patrón de herencia paradominante a causa de la frecuencia de hemangiomas en varios familiares afectados.6,7
Baskerville y cols.,8 informaron los resultados de un estudio que utilizó flebografía; en el que se observó estenosis venosa profunda en solo cinco de las 36 extremidades afectadas. En consecuencia, el autor argumentó que el SKT es un trastorno mesodérmico y que las anomalías venosas profundas no son parte de su etiología, sino una de sus numerosas características clínicas.
Se diagnóstica, en su mayoría durante la infancia por la presencia de dos de los tres rasgos clínicos anteriores y las pruebas de imagen. Por la baja frecuencia de presentación en la adultez se decide describir los hallazgos clínicos, de laboratorio, de imágenes y la evolución del siguiente caso.
PRESENTACIÓN DEL CASO
Paciente femenina de 31 años, mestiza, que acude al servicio de Urgencia del Hospital “Dr. Salvador Allende”, con antecedente patológicos personales de diabetes mellitus tipo 2, insuficiencia venosa profunda y úlcera varicosa a repetición desde hace nueve años. Cursaba con cuadro clínico más o menos de una semana de evolución, caracterizado por edema en miembro inferior izquierdo asociado a dolor y limitación funcional. Además de sangramiento rectal.
Al examen físico se observa asimetría de ambos miembros inferiores con hipertrofia del izquierdo y moderada cianosis a ese nivel. Se nota presencia de várices en la región lateral del muslo y a la palpación ausencia de pulsos poplíteo y pedio de la extremidad afecta con pérdida de sensibilidad superficial y profunda. Dentro de los exámenes de laboratorio se realizaron biometría hemática completa, electrolitos, enzimas hepáticas y cardiacas, parámetros nutricionales, coagulograma, PCR cuantitativa y procalcitonina, los mismos que se encontraron dentro de los rangos normales.
Se valora por el servicio de Angiología, quienes solicitan un estudio de angiotomografía. En esta se identifican la vena cava inferior y la arteria aorta abdominal al unísono, al tener la vena cava inferior el aspecto de una arteria de gran calibre (arterialización) además de la circulación distal (arteria tibial posterior). (Fig. 1)
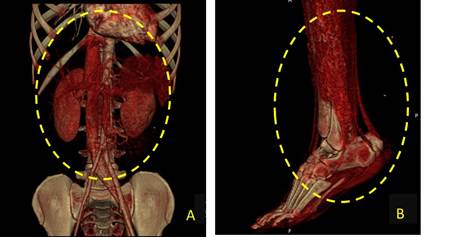
Fig. 1 Angiotomografía donde se observan la vena cava inferior y la arteria aorta abdominal al unísono (A) y la circulación distal (B).
Las imágenes procesadas en in space muestran la existencia de gran cantidad de fístulas arteriovenosas en el miembro inferior izquierdo, con la identificación paralela de los gruesos vasos (arteria y vena) distales al mismo tiempo, por lo que se corrobora la presencia de un Síndrome Klippel- Trénaunay. (Fig. 2)
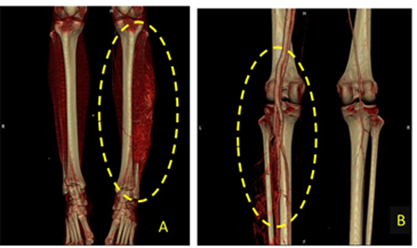
Fig. 2 Angiotomografía donde se observa la existencia de gran cantidad de fístulas arteriovenosas (A) en el miembro inferior izquierdo con identificación paralela de los gruesos vasos (arteria y vena) (B)
Recibe tratamiento con escleroterapia y terapia compresiva, logra reducir en más de la mitad el aumento de volumen del miembro inferior izquierdo y la rectorragia, con evolución satisfactoria ante los medios de acción aplicados. Las pruebas de laboratorio clínico y especiales se mantienen en la actualidad, dentro de límites normales y continua su seguimiento por consulta externa de cirugía vascular.
DISCUSIÓN
El Síndrome de Klippel y Trénaunay mantiene una incidencia similar en ambos sexos y se presenta desde el nacimiento, con dos o más de sus síntomas y signos correspondientes a la triada que lo caracteriza. No obstante, el diagnóstico puede realizarse de forma tardía según las manifestaciones clínicas y la edad en la que comienzan a desarrollar trastornos más severos al individuo.9,10 Este caso se tuvo un diagnóstico tardío, de ahí las complicaciones, para su análisis se seleccionó la angiotomografía, por ser una herramienta con alta especificidad y sensibilidad en estos casos.
Afecta las extremidades inferiores de forma unilateral (95 %), se han descrito con menos frecuencia en ambas extremidades inferiores, en las extremidades superiores (5 %) uni o bilateralmente, en las cuatro extremidades de forma simultánea, y cuadros limitados al tórax, pelvis, abdomen, cabeza o cuello. Las estructuras intraabdominales y torácicas se afectan con menos frecuencia.5 Como se muestra en esta investigación el caso tenía afectaciones abdominales y en el miembro inferior izquierdo.
La malformación capilar, también conocida como mancha en vino de Oporto, nevo telangiectásico o angioma plano, es la manifestación cutánea más frecuente, presente ya en el nacimiento, se asocia a una mayor frecuencia de malformaciones linfáticas y complicaciones como infecciones, sangrado o bien definido cuya intensidad de coloración puede aumentar con la edad.11
Se coincide con el informe de Marunraj y cols.,11 quienes plantean que las malformaciones venosas no están siempre presentes en el nacimiento. Se manifiestan durante la bipedestación y pueden presentarse como anomalías del sistema venoso superficial, profundo o de cualquier vena, incluso la cava inferior, o venas embrionarias persistentes. Estas son varicosas, atípicas del sistema venoso superficial y corresponden a la vena lateral del muslo o la vena ciática. El caso que se discute tenía 31 años de edad y hacía algún tiempo ya tenía las várices, pero nunca se le había sido diagnosticado dicho síndrome.
Suelen ser venas largas y tortuosas que pueden carecer de válvulas y causar síntomas de pesadez en las extremidades inferiores. Entre las anormalidades en el sistema superficial, es posible encontrar desde ectasia de pequeñas venas y varicosidades hasta grandes malformaciones venosas. Las alteraciones en el sistema venoso profundo incluyen dilataciones aneurismáticas, duplicaciones, hipoplasia, aplasia y compresión externa por vasos anómalos o bandas fibróticas. Las venas poplíteas y femorales superficiales son afectadas con más frecuencia.12 En el examen físico no se detectaron pulsos poplíteo y pedio con pérdida de sensibilidad tanto superficial como profunda.
Algunos pacientes también pueden presentar venas varicosas perianales y perirrectales, producto a un alto flujo en la vena ilíaca interna,13 como es el presente caso donde la paciente manifestó rectorragia. En ocasiones, las malformaciones venosas pueden extenderse al mediastino posterior y al espacio retropleural, aunque rara vez producen síntomas.13
Las complicaciones que se pueden asociar son: la hemorragia, la tromboflebitis (25-50 %), la presencia de comunicaciones arteriovenosas, que pueden acompañarse de fallo cardíaco congestivo por alto flujo, y el tromboembolismo pulmonar (5-30 %). Las complicaciones pelvianas van desde la presencia de hematuria, hemorragia gastrointestinal y estreñimiento, hasta la obstrucción del meato vesical y las infecciones recurrentes por la microbiota intestinal. Las malformaciones linfáticas pueden deberse a una hipoplasia linfática, presente en más del 55 % de los pacientes, y asociarse a linfedema o macroquistes linfáticos aislados en la pelvis y las extremidades.14
El diagnóstico diferencial del Síndrome Klippel y Trénaunay se establece con otra serie de síndromes que comparten alguno o algunos de sus rasgos clínicos, entre los que se menciona a los síndromes: de Proteus, Parkes Weber, Bannayan-Riley-Ruvalcaba, Maffuci, entre otros.11
El tratamiento del Síndrome Klippel y Trénaunay es multidisciplinario, está destinado a mejorar la función y calidad de vida (epifisiodesis endoscópica, amputación de los dedos), prevenir y tratar las complicaciones (antibióticos y anticoagulantes, medias de compresión) y, en la medida de lo posible, su aspecto (escleroterapia, tratamiento con láser). 14 En esta paciente se utilizó la terapia compresiva y la escleroterapia para el tratamiento, con resultados satisfactorios.
El síndrome de Klippel y Trénaunay tiene un patrón clínico e imagenológico característico que, en su conjunto, son concluyentes en el diagnóstico. De esta forma se alcanza un adecuado diagnóstico, seguimiento y tratamiento médico por el equipo multidisciplinario responsable de la atención de estos pacientes, se logra así una mejor calidad de vida

