










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta Médica Espirituana
versión On-line ISSN 1608-8921
Gac Méd Espirit vol.17 no.3 Sancti Spíritus dic. 2015
PRESENTACIÓN DE CASO
Síndrome Waardenburg. Presentación de una familia afectada
Syndrome Waardenburg. Presentation of an affected family
Dra. Elayne Esther Santana HernándezI, Dr. Victor Jesús Tamayo ChangII.
I Centro Provincial Genética Holguín. Cuba.
II Hospital Pediátrico Universitario Octavio de la Concepción de la Pedraja. Holguín.Cuba.
RESUMEN
Fundamento: El síndrome de Waardenburg es una enfermedad genética caracterizada por anomalías de la pigmentación y sordera neurosensorial. Se describen varios tipos clínicos, con gran heterogeneidad genética, la mayoría de los casos publicados presentan un patrón de herencia autosómico dominante, aunque se describen otras formas de herencia.
Objetivo: Mostrar una familia con varios miembros afectados representativa de expresividad variable.
Presentación de caso: Se presenta una familia con siete enfermos donde predomina la hipoacusia de grado variable al igual que las alteraciones de la pigmentación de la piel, el pelo y el iris. El adecuado diagnóstico y asesoramiento genético, unido a la oportuna intervención con el implante coclear ha permitido la incorporación adecuada en la enseñanza normal a estos enfermos.
Conclusiones: Es importante el diagnóstico oportuno para realizar acciones con la finalidad de mejorar la calidad de vida y la correcta incorporación social de estos pacientes.
Palabras clave: Síndrome de Waardenburg; genética; familia.
DeCS: SÍNDROME DE WAARDENBURG/genética; FAMILIA.
ABSTRACT
Background: The syndrome of Waardenburg is a genetic illness characterized by anomalies of the pigmentation and neurosensory deafness. Several clinical types are described, with great genetic heterogeneity; most of the published cases present a pattern of autos’omico dominant inheritance, although other inheritance forms are described
Objective: To show a representative family with several affected members of variable expression.
Case presentation: A family is presented with seven sick persons where the hypo acoustic of variable grade prevail the same as the alterations of the pigmentation of the skin, the hair and the iris. The appropriate diagnose and genetic advice, together to the opportune intervention with the cochlear implants has allowed the adapted incorporation in the normal teaching to these sick persons
Conclusions: It is important the opportune diagnosis to carry out actions with the purpose of improving the quality of life and the correct social incorporation of these patients.
Keywords: Síndrome de Waardenburg; genética; familia.
MeSH: WAARDENBURG SYNDROME/genetics; FAMILY.
INTRODUCCIÓN
El síndrome de Waardenburg (SW) fue descrito desde 1951, sus características físicas reportadas fueron, sordera neurosensorial congénita unilateral o bilateral, desplazamiento lateral del canto interno de los ojos y del conducto lagrimal inferior, raíz nasal alta y amplia, heterocromía del iris, sinofris, hipertricosis del tercio medio de las pestañas 1,2.
Inicialmente de describieron cuatro formas clínicas, pero otros investigadores demostraron que hay heterogeneidad genética en este síndrome y lo clasificaron inicialmente en dos formas, basándose en si la distopia cantorum está presente (Tipo 1), o ausente (Tipo 2). Un tercer tipo que se caracteriza por ptosis unilateral, sin distopia cantorum, y el tipo 4 asociado enfermedad de Hirschprung 3-5.
El síndrome de Waardenburg tipo 1 y 2, muestra un patrón de herencia autosómico dominante con una expresiva variables. La tasa de mutaciones es del 0.4 x 10E-4 por gameto, y una avanzada edad paterna parece ser un factor de riesgo en los casos de mutaciones nuevas. Se estima que uno de 10.000 personas porta el gen que causa este síndrome 4.
Este síndrome está causado por una migración o diferenciación anormal de células de la cresta neuronal durante el desarrollo embrionario 6-8. Se describe que en periodo neonatal, los pacientes presentan anomalías en la pigmentación (mechón blanco, cejas y pestañas blancas, parches blancos en la piel y heterocromía del iris) asociadas a una obstrucción intestinal 7. La sordera neurosensorial es frecuente, precoz, y puede ser unilateral. El desarrollo psicomotor es normal y no presentan discapacidad intelectual, de ahí la importancia de la rehabilitación auditiva precoz 9,10.
PRESENTACIÓN DE CASO
PRESENTACIÓN DE LA FAMILIA
Se presenta una familia de cuatro generaciones, donde en todas las generaciones, cada uno de estos con expresividad variable en cuanto a la hipoacusia y el piebaldismo, lo que esta descrito en esta afección y dificulta en ocasiones su diagnóstico lo cual entorpece la intervención precoz por parte de los audiólogos para que se evalué tempranamente el implante coclear para la mejor y adecuada inserción educativa y social de estos afectados.
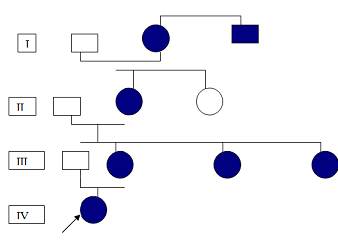
Después de aceptar participar en el estudio firmando consentimiento infirmado, se les realiza examen físico completo oftalmológico y de audiología a varios miembros de la familia, diagnosticándose sietes afectados con el síndrome de Waardenburg, todos con expresión fenotípica diferente, con gran heterogeneidad clínica, donde solo en la menor de la última generación, se aprecia expresión severa de la enfermedad. Se presentan afectados en las cuatro generaciones y todos los afectados tienen como mínimo un progenitor afectado, mostrando esta familia un patrón de herencia autosómico dominante, como se puede apreciar en la figura 1.
Figura 1. Árbol genealógico de la familia afectada.
Esta paciente con síndrome Waardenburg tipo 1, presenta hipoacusia bilateral profunda, mancha hipopigmentada la pierna izquierda (piebaldismo) asociado a heterocromía del iris en este caso un ojo azul el derecho y el ojo izquierdo pardo, “distopia cantorum” (desplazamiento lateral del canto interno) como se puede apreciar en la figura 2.
Figura 2. Características fenotípicas faciales.

Estas mismas alteraciones de encontraron en los siete afectados pero en grado variable donde la hipoacusia fue bilateral profunda solo en tres pacientes, dos hipoacusia unilateral asociado a heterocromía del iris y otros dos con la hipoacusia moderada y el piebaldismo, en estos seis casos anteriores no se realizó diagnóstico oportuno, en esta afectada se realizó el diagnóstico a los dos años de edad, con un buen desarrollo pondoestatural y sicomotor. Se le operó de inmediato y se le hizo el implante coclear para que desarrollara su lenguaje, y su incorporación escolar y social fuera la más adecuada posible. Actualmente se encuentra en una enseñanza normal correspondiente con su edad cronológica aunque continúa con su rehabilitación por logofoniatría.
DISCUSIÓN
El síndrome de Waardenburg se clasifica en diferentes grados o tipos, según los síntomas que presente el paciente:
Tipo 1: (Gen PAX3, locus 2q35). Característico la llamada “distopia cantorum” (desplazamiento lateral del canto interno). Sordera neurosensorial congénita. Heterocromía del iris tanto completa como segmental o un iris azul intenso. Pigmentación del cabello, blanco en cejas y pestañas y mechón blanco, además de canas prematuras1-3.
Tipo 2: (Gen MITF, locus 3p14.1-p12.3). Sordera neurosensorial congénita. Anormalidades en la pigmentación del iris, segmental, completa o azul intenso. Pigmentación del cabello, también blanco en cejas y pestañas y mechón blanco, además de canas prematuras. Encontramos dos subclases dentro de este: Subtipo A: Paladar hendido, espina bífida, vagina sin desarrollar. Subtipo B: Ausencia de afección sistémica 2.
Tipo 3: (Gen PAX3). Menos común; afecta las extremidades, impidiendo una completa flexión, deformidades o sindactilia (fusión de varios dedos) 2.
Tipo 4: (Genes: EDN3, EDNRB y SOX10). Es muy raro y surge de la asociación de un síndrome de Waardenburg de tipo 2 con la enfermedad de Hirschsprung, siendo esta enfermedad un trastorno congénito. Afecciones graves neurológicas. Se han identificado tres genes responsables de esta enfermedad: el gen EDNRB (13q22.3), que codifica para el receptor de la endotelina B, el gen EDN3 (20q13.32), que codifica para un ligando del receptor de la endotelina y el gen SOX10 (22q13.1), que codifica para el factor de trascripción SOX 10 3.
El tipo 1, ligado al locus ABO sobre el cromosoma 9 (9q34) y también existe un gen localizado en 2q35 q37. Otras investigaciones han demostrado ligamiento del SW con la fosfatasa alcalina, la que estaba previamente asignada a 2q37. Los estudios más recientes han demostrado ligamento entre este síndrome y los marcadores PAX 3 MITF. Las mutaciones en el gen SOX10 se trasmiten de manera autosómica dominante 3-5.
El caso integra una familia en la que hay siete miembros afectados con SW tipo 1 distribuidos en cuatro generaciones; en esta familia se observa la penetrancia completa y expresividad variable de la enfermedad.
El síndrome de Waardenburg tipo 1 (SW1), caracterizado por sordera o hipoacusia, distopia cantorum, alteraciones pigmentarias del iris (heterocromía total del iris, o parcial o isohipocromía (ambos ojos azules, pálidos o poco brillantes) y el cabello. Más del 90 % de los individuos tienen mutaciones en el gen PAX3, lo cual evidencia la heterogeneidad de locus 6,7. El modo de herencia es autosómico dominante con una penetrancia reducida, expresividad variable y acentuada heterogeneidad genética. Otros diagnósticos que se deben tener en cuenta para diferenciar son: el vitíligo, albinismo total y el piebaldismo aislado sin hipoacusia, el síndrome de Teitz (hipopigmentación generalizada con sordera congénita), otros tipos de hipoacusia, los se despreciaron al realizarle el examen oftalmológico y audiológico 8-10.
CONCLUSIONES
En esta familia se trasmitió la enfermedad con un patrón de herencia autosómico dominante, con una alta penetrancia observándose en cuatro generaciones, individuos afectados con gran heterogeneidad clínica y expresividad variable, esto nos hace suponer que entre ellos debe existir también heterogeneidad genética alélica o no alélica de locus.
REFERENCIAS BIBLIOGRÁFICAS
1. Demirci GT, Atis G, Altunay IK. Waardenburg Syndrome type 1: A case report. Dermatol Online J [Internet]. 2011 Nov [cited: 2015 Ene];17(11):3. Available from: http://escholarship.org/uc/item/14k128r2
2. Zaman A, Capper R, Baddoo W. Waardenburg Syndrome: More common than you think. Clin Otolaryngol [Internet]. 2015 Feb [cited: 2015 Ene]; 40(1):44-8. Available from: http://onlinelibrary.wiley.com/doi/10.1111/coa.12312/abstract;jsessionid=7C7248A9EABC15B
3. Imperato PJ, Imperato GH. Clinical Manifestations of Waardenburg Syndrome in a Male Adolescent in Mali, West Africa. J Community Health [Internet]. 2015 Feb [cited Ene 2015] ;40(1):103-9. Available from: http://download.springer.com/static/pdf/355/art%253A10.1007%252Fs10900-014-9942-7.pdf?originUrl=http%3A%2F%2Flink.springer.com%2Farticle%2F10.1007%2Fs10900-014-9942-7&token2=exp=1442417981~acl=%2Fstatic%2Fpdf%2F355%2Fart%25253A10.1007%25252Fs10900-014-9942-7.pdf%3ForiginUrl%3Dhttp%253A%252F%252Flink.springer.com%252Farticle%252F10.1007%252Fs10900-014-9942-7*~hmac=696b1f065312e62d4cdcfaf74316ab4edc7a991d51a4024ce14903fa328d264b
4. Chen K, Zong L, Liu M, Zhan Y, Wu X, Zou W, Jiang H. De novo dominant mutation of SOX10 gene in a Chinese family with Waardenburg syndrome type II. Int J Pediatr Otorhinolaryngol [Internet]. 2014 Jun [cited Ene 2015];78(6):926-9. Available from: http://www.ijporlonline.com/article/S0165-5876%2814%2900146-3/abstract
5. Jelena B, Christina L, Eric V, Fabiola QR. Phenotypic variability in Waardenburg syndrome resulting from a 22q12.3-q13.1 microdeletion involving SOX10. Am J Med Genet A [Internet]. 2014 Jun [cited 2014 Ago];164A(6):1512-9. Available from: http://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.36446/epdf
6. Mahmoudi A, Rami M, Khattala K, Elmadi A, Afifi MA, Youssef B. Shah-Waardenburg syndrome. Pan Afr Med J [Internet]. 2013 [2015 Ene];14:60. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3617621/
7. Wildhardt G, Zirn B, Graul-Neumann LM, Wechtenbruch J, Suckfüll M, Buske A, et al. Spectrum of novel mutations found in Waardenburg syndrome types 1 and 2: implications for molecular genetic diagnostics. BMJ Open [Internet]. 2013 Mar 18 [cited: 2015 Ene];3(3):pii: e001917. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3612789/
8. Nasser LS, Paranaíba LM, Frota AC, Gomes A, Versiani G, Martelli Júnior H. Waardenburg syndrome--ophthalmic findings and criteria for diagnosis: case reports. Arq Bras Oftalmol [Internet]. 2012 Oct [cited: 2015 Ene];75(5):352-5. Available from: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-27492012000500012&lng=en&nrm=iso&tlng=en
9. Fernández RM, Núñez-Ramos R, Enguix-Riego MV, Román-Rodríguez FJ, Galán-Gómez E, et al. Waardenburg syndrome type 4: report of two new cases caused by SOX10 mutations in Spain. Am J Med Genet A [Internet]. 2014 Feb [cited: 2015 Ene];164A(2):542-7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/?term=Waardenburg+syndrome+type+4%3A+report+of+two+new+cases+caused+by+SOX10+mutations+in+Spain.+Am+J+Med+Genet+A
10. Traoré H, Traoré D, Ouane O, Simpara B, Ongoiba N. A case report on Waardenburg syndrome with cleft lip. Mali Med [Internet]. 2011 [cited: 2015 Ene];26(3):53-5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22766441
Recibido: 2015-05-28
Aprobado: 2015-11-09
Dra.Elayne Esther Santana Hernandez. Centro Provincial Genética Holguín. Cuba

