










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev. Med. Electrón. v.31 n.1 Matanzas ene.-feb. 2009
POLICLÍNICO DOCENTE RAMÓN MARTÍNEZ. VARADERO
Síndrome de Ehlers-Danlos. Presentación de un caso
Ehlers-Danlos syndrome. Presentation of a case
AUTORAS
Dra. Milaydi Moreira González. (1)
E-mail: genetica.mtz@infomed.sld.cu
Dra. Hilda Álvarez de la Campa Gil. (1)
Dra. Dairys Laura Falcón Rodríguez. (2)
Lic.Claribel Lugo Rodríguez.(3)(1)Especialista de 1er. Grado en Medicina General Integral. Máster en Asesoramiento Genético. Policlínico Docente Dr."Ramón Martínez". Varadero.
(2)Especialista de 1er. Grado en Genética Clínica. Centro de Genética Provincial .Matanzas.
(3)Máster en Asesoramiento Genético.Licenciada en Enfermería. Policlínico Docente Dr."Ramón Martínez". Varadero.RESUMEN
En este trabajo se realizó la presentación de un caso afectado con el Síndrome de Ehlers- Danlos, identificado en la consulta de Genética Clínica. Teniendo en cuenta la importancia y severidad de algunas de sus formas, y que el diagnóstico, en ocasiones, de las formas más leves no se realiza, se revisó la bibliográfica actualizada sobre esta patología para la mejor comprensión del caso.
DeCS:
SÍNDROME DE EHLERS-DANLOS/genética
HUMANOS
MASCULINO
PREESCOLARINTRODUCCIÓN
El Síndrome de Ehlers-Danlos (SED) es una enfermedad hereditaria del colágeno que se presenta sobre todo como desórdenes dermatológicos, hipermovilidad de las articulaciones, entre otros. (1-3) El SED se caracteriza por excesiva movilidad de las articulaciones, piel hiperelástica, frágil y que presenta equimosis con gran facilidad, vasos sanguíneos que se deterioran fácilmente y en pocas ocasiones ruptura de órganos internos. (3,4) Este síndrome implica la formación anormal de tejido conectivo y ocurre genéticamente de diferentes formas. Los síntomas van desde leves hasta severos, lo que dificulta el reconocimiento clínico de la enfermedad. En Cuba no existe registro de la incidencia o prevalencia del SED, aunque sí se diagnostican las formas clásicas y las severas, escapando en ocasiones las formas leves al diagnóstico, lo cual está influenciado en gran medida por la heterogeneidad genética y la variabilidad clínica del mismo.(5-7)
PRESENTACIÓN DEL CASO
Descripción clínica: Paciente masculino, raza blanca, de 3 años de edad, que es remitido por ortopedia por presentar tendencia a las luxaciones, articulaciones hiperextensibles y piel hiperelástica, fina, que se daña con facilidad. Al interrogatorio se recogen los antecedentes patológicos familiares, donde se refiere que la abuela materna falleció a la edad de 55 años, de una muerte súbita, demostrándose en los hallazgos de la necropsia que la causa fue la rotura de un aneurisma disecante de la aorta. La madre del afectado tiene diagnosticado un SED tipo I, además ésta refiere que su madre había tenido una muerte fetal aproximadamente a las 26 semanas de gestación. Por la vía paterna no se recogen antecedentes de interés. Al indagar sobre los antecedentes obstétricos se plantea que fue un parto pretérmino, con una edad gestacional de 35 semanas, con rotura prematura de membranas. Entre los antecedentes postnatales del niño llama la atención que hubo un desarrollo motor lento, tendencia a los traumas y dificultad en la cicatrización. Al examen físico: Cara: puente nasal ancho y pliegues epicánticos, Orejas hipermóviles, SOMA: talla disminuida, cuello largo, pies planos, articulaciones hiperextensibles, genus recurvatun, escoliosis. Piel: fina, hiperelástica, cicatrices en papel de cigarro. Luego de realizar el interrogatorio y el examen físico, se procedió a confeccionar el árbol genealógico que quedó conformado de la siguiente forma:
ÁRBOL GENEALÓGICO
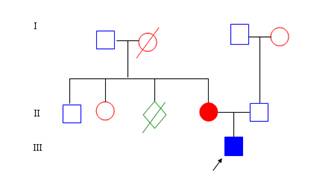
Se realiza la entrevista, ésta incluye el interrogatorio a los padres, el examen físico al paciente y los estudios específicos correspondientes. Se confecciona la historia clínica genética. Se le realiza un examen físico exhaustivo , donde se comprueban signos y características clínicas sugestivas de un SED tipo I.
Se les informa a los padres del diagnóstico presuntivo y la necesidad de realizar algunos exámenes, como el examen de agregación plaquetaria y la valoración por oftalmología y cardiología para descartar otras afectaciones. Se brinda apoyo psicológico a esta pareja. En los exámenes realizados se confirma el diagnóstico de la enfermedad, donde se informa un trastorno de la agregación plaquetaria con ADP. En la valoración oftalmológica sólo se detecta un trastorno de refracción (miopía) y el examen cardiovascular resultó ser negativo. Se explica sobre las manifestaciones clínicas fundamentales de la enfermedad como son la hiperextensibilidad de las articulaciones que llevan a luxaciones frecuentas, por lo cual se deben evitar los movimientos bruscos y las situaciones que conllevan a estas complicaciones, además evitar las heridas y traumatismos debido a los trastornos de la cicatrización a que tienden estos pacientes, así como evitar los procederes quirúrgicos, realizándose sólo los imprescindibles.
Se descartan otros tipos de SED como son el tipo III que, aunque predominan la marcada laxitud de las articulaciones acompañada por luxaciones frecuentes, la piel es generalmente de carácter normal. En el tipo IV predominan los trastornos vasculares y las varicosidades venosas son comunes y llegan a ser severas, y tiene un patrón de herencia AD y AR. El tipo V tiene un patrón de herencia recesiva ligada al X, descartándose ésta ya que en la familia afectada los pacientes afectados son de ambos sexos.
El SED tipo VI escoliótico-ocular tiene una herencia AR, sus características principales son hipotonía muy marcada en el período de recién nacido y escoliosis progresiva, la cual puede aparecer tempranamente en la infancia. Si la escoliosis es severa, el compromiso cardiopulmonar puede acortar el tiempo de vida. En el SED tipo VII, Artrocalasia múltiple congénita, VIIA y VIIB hay dislocación bilateral congénita de cadera y piel relativamente normal, los individuos afectados tienen dificultades con la movilidad de las articulaciones que les limita deambular, mientras otros logran recuperarse o deambulan bien mediante cirugía. El SED TIPO VII DERMATOSPARAXIS tiene herencia AR, hay una sorprendente laxitud de las articulaciones, piel extremadamente suave, frágil y extensible, daños fáciles, hirsutismo ligero, esclerótica azul, también se puede encontrar retraso del crecimiento, miembros cortos, hernia umbilical y rasgos faciales característicos con micrognatia, párpados prominentes e hinchados, lo cual no se constata en este paciente. En el SED TIPO VIII PERIODONTAL se presenta facilidad para los hematomas, fragilidad cutánea moderada, cicatrización anómala y periodontitis grave con pérdida prematura de los dientes y del hueso alveolar, no estando esto presente en el paciente. El SED TIPO IX. ESQUELÉTICO RECESIVO LIGADO AL CROMOSOMA X se caracteriza por piel suave, hiperextensible, exostosis occipital, ensanchamiento y arqueamiento de los huesos largos en las zonas de inserción ligamentosa y tendinosa, clavículas anchas y cortas, diarrea crónica, divertículo de vejiga con roturas espontáneas, y hernias inguinal. El SED tipo X. Hay un defecto de fibronectina, que se corrige con su administración, hay facilidad para la formación de hematomas. En el caso del SED tipo II, es el que junto al Tipo I forman los tipos clásicos, pero en el caso del II es mucho más ligero. Por lo anteriormente expuesto se hace el diagnóstico clínico del paciente, descartándose unas formas por su tipo de herencia y en otros casos por no estar presentes los signos característicos de cada una de ellas.
Se le informa con relación al pronóstico de la enfermedad que generalmente tienen un período de vida normal, debiendo tomar las precauciones necesarias para evitar la aparición de complicaciones, además se sugiere realizar ecografías periódicas para diagnosticar precozmente la formación de aneurismas. Se explica también que hay manifestaciones del SED que pueden aparecer en cualquier etapa de la vida, como son las afectaciones de las válvulas cardíacas, entre otras, por lo que se requiere de un seguimiento estrecho, pues esto implicaría tomar medidas profilácticas ante determinadas situaciones como las extracciones dentarias evitando así la endocarditis bacteriana. Se orienta el seguimiento periódico por las especialidades médicas correspondientes, así como un chequeo médico una vez al año.
Se explica sobre el patrón de herencia del SED tipo I, que es autosómica dominante, siendo mapeados en la actualidad genes en los siguientes locus ---- 17q21.31-q32, 9q34.2-q34.3, 2q31. Las mutaciones subyacentes parecen estar en los tipos V de genes colágenos, COL5A1 y COL5A2. En este tipo de herencia hay una probabilidad de estar afectada el 50 % de su descendencia para cada embarazo futuro, siendo esta probabilidad igual para cada embarazo. Se brinda apoyo psicológico y orientación a la familia.REFERENCIAS BIBLIOGRÁFICAS
1. Letorneau Y, Perusse R, Buithieu H. Manifestaciones Orales del Síndrome de Ehler-Danlos;2003. Disponible en: http://www.ceda/spanish/oral.htm.
2. Kobayasi T. Dermal Elastic Fibres in the Inherited Hypermobile Disorders. J Dermatol Sci. 2005; 30.
3. Brooks DG. Síndrome de Ehler-Danlos;2001. Disponible en:http://www.nlm.nih.gov/medlineplus/Spanish/ency/article/001468.htm.
4. Lawrence EJ. The Clinical Presentation of Ehlers-Danlos Syndrome. Adv Neonatal Care. 2005 Dec; 5(6):301-14.
5. Hamel BC. Ehle rs-Danlos Syndrome. Neth J Med. 2004; 62(5):140-2.
6. Uitto J. The Ehlers-Danlos Syndrome Phenotypic Spectrum and Molecular Genetics. Eur J Dermatol. 2005; 15(5):311-2
7. Lacadena JR. Asesoramiento Genético. La toma de decisión. Aspectos Éticos;2000. Disponible en: http://www.google.com.
8. Penchaszadeh VB, Puñales Morejón D. Dimensiones psicosociales de los problemas genéticos. USA: División de Genética Médica Beth Israel Medical Center;2000.
9. Pérez ET. Metodología del Asesoramiento Genético. La Habana:CNGM; 2003.
10. Rojas BI. Principios del Asesoramiento Genético. La Habana: CNGM; 2003.
11. Harper PS. Practical Genetic Counselling. 5thed. HPS(eds).Boston:Butternorth Heinemann;1999.SUMMARY
We present a case of a patient affected by the Ehlers-Danlos syndrome, identified at the Genetic Clinic consultation. Taking into account the importance and severity of some of its forms, and that sometimes the mildest forms are not diagnosed, we carried out an up-dated bibliographic review on this pathology for a better comprehension of the case.
MeSH:
EHLERS- DANLOS SYNDROME/genetics
HUMANS
MALE
CHILD,PRESCHOOLCÓMO CITAR ESTE ARTÍCULO
Moreira González M, Álvarez de la Campa Gil H , Falcón Rodríguez DL, Lugo Rodríguez C. Síndrome de Ehlers-Danlos. Presentación de un caso. Rev méd electrón[Seriada en línea] 2009; 31(1). Disponible en URL: http://www.revmatanzas.sld.cu/revista%20medica/ano%202009/vol1%202009/tema13.htm [consulta: fecha de acceso]

