










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev. Med. Electrón. v.32 n.5 Matanzas sep.-oct. 2010
HOSPITAL PROVINCIAL DOCENTE CLÍNICO QUIRÚRGICO JOSÉ RAMÓN LÓPEZ TABRANE. MATANZAS
Revista Médica Electrónica. 2010;32(5)
Malformación de Arnold Chiari tipo I. Presentación de un caso
Arnold-Chiari malformation Type I. Presentation of a case
AUTORES
Dr. Ihosvany Ruiz Hernández (1)
Est. Antonis Cano Soler (2)
1) Especialista de II Grado en Medicina Interna. Máster en Infectología. Profesor Auxiliar. Hospital Provincial Docente Clínico Quirúrgico José Ramón López Tabrane. Matanzas.
2) Estudiante de 4to año de la carrera de Medicina. Alumno ayudante de Cirugía. Hospital Provincial Docente Clínico Quirúrgico José Ramón López Tabrane. Matanzas.
RESUMEN
La malformación de Arnold Chiari es una rara enfermedad incapacitante que afecta al 0,5 % de la población, siendo el 80 % mujeres. La hidrocefalia resultante es causada por el exceso de líquido cefalo raquídeo en el sistema ventricular, o por una falta de equilibrio entre la formación y su absorción, lo que da lugar a un progresivo aumento de las cavidades ventriculares. Se presenta el caso de una paciente con malformación de Arnold Chiari tipo I, en la cual hay un descenso anormal en grado variable de la porción inferior del cerebelo, del bulbo y el cuarto ventrículo hacia el conducto raquídeo a través del agujero occipital. El objetivo de este trabajo es describir el cuadro clínico y compararlo con la literatura médica revisada, del caso en cuestión.
DeCS
TRASTORNOS DE CEFALALGIA/diagnóstico
MALFORMACIÓN DE ARNOLD-CHIARI/diagnóstico
MALFORMACIÓN DE ARNOLD-CHIARI/complicaciones
MALFORMACIÓN DE ARNOLD-CHIARI/quimioterapia
MALFORMACIÓN DE ARNOLD-CHIARI/cirugía
HIDROCEFALIA/etiología
INFORMES DE CASOS
REVISIÓN
HUMANOS
FEMENINO
ADULTO
INTRODUCCIÓN
El síndrome de Arnold Chiari es una malformación rara y congénita del sistema nervioso central, que consiste en un descenso del cerebelo introduciéndose este en el canal medular. Está localizada en la fosa posterior o base del cerebro y pertenece al grupo de las malformaciones craneocervicales o alteraciones de la charnela (1).
Una de las situaciones más traumáticas de los afectados por esta enfermedad aparece cuando muestran inestabilidad en la charnela y deben ser intervenidos quirúrgicamente para fijarles toda la articulación del cuello, por lo que deben llevar durante un año un halo en la cabeza. Esta situación provoca estados de angustia en los pacientes, ya que por el desconocimiento de la sociedad sobre su enfermedad se sienten observados.
El síndrome de Chiari puede ser sintomático o asintomático. Una vez que aparecen los síntomas se considera que ya existe enfermedad. Existen dos tipos de síndrome de Arnold Chiari: el síndrome de Chiari tipo I o de adulto, y el síndrome Chiari tipo II o infantil (que está asociado a meningocele y a veces a espina bífida) (2).
Esta patología puede llegar a producir diferentes síntomas, todos ellos causados por los siguientes motivos:
-Al encontrarse la hernia del cerebelo en el canal medular, produce una presión en todas las terminaciones nerviosas causando varios síntomas.
-Este descenso del cerebelo hace de tapón de tal forma que el líquido cefaloraquídeo (LCR) puede producir dos enfermedades secundarias como son: la hidrocefalia (acúmulo de LCR en la cabeza) y siringomielia (acúmulo de LCR en la médula), generando quistes siringomiélicos. El 55 % de los pacientes afectados por el síndrome de Chiari tienen siringomielia, dolencia que atrofia los músculos y produce alteraciones en la sensibilidad (2).
La patología de Arnold Chiari es una enfermedad de carácter progresivo, calificada como enfermedad rara dada su baja morbilidad en la población en general que, fundamentalmente, actúa sobre el sistema nervioso central, deteriorando, como consecuencia de ello, la calidad de vida de los pacientes si no es diagnosticada precozmente y si no es aplicada la técnica quirúrgica adecuada (3).
Esta patología del sistema nervioso central, provoca más de 100 síntomas asociados, es causa de minusvalía y uno de los principales motivos de fallecimiento de los neonatos con espina bífida (4).
Bajo este término se describen una serie de anormalidades congénitas de la base cerebral, entre las más consistentes se hallan (5):
• Extensión de una lengüeta de tejido cerebeloso, posterior al bulbo y médula, en la región del canal cervical.
• Desplazamiento del bulbo en el canal cervical con la porción inferior del cuarto ventrículo.
En los últimos años, el término de malformación de Arnol-Chiari se ha restringido a los tipos I y II de Chiari, esto es la malformación cerebelobulbar con o sin mielomeningocele respectivamente. El tipo III no es más que un meningomielocele cervical alto u occipitocervical con herniación tonsilar, mientras que el IV consiste sólo en hipoplasia cerebelosa (6).
El bulbo y el puente están alongados, y el acueducto estrechado. Los tejidos desplazados (bulbo y cerebelo) ocluyen el foramen magnum; y el cerebelo restante, que es pequeño, es también desplazado obliterando la cisterna magna. Los agujeros de Luschka y Magendie se abren en el canal cervical, y el tejido aracnoideo alrededor del tronco y cerebelo herniado es fibrótico. Todos estos factores son operativos en la producción de hidrocefalia, la cual puede estar presente. En este tipo de malformación un mielo meningocele casi siempre se asocia y la hidromielia del canal cervical es también común (7,8).
Anormalidades del desarrollo del cerebro (particularmente polimicrogiria) pueden coexistir, al tiempo que el extremo final de la médula espinal (filium terminal) puede extenderse tan bajo como el sacro. La fosa posterior es pequeña, el foramem magnum está agrandado. La base del cráneo está aplanada o hay impresión basilar (9). Los autores de este trabajo se proponen presentar un caso con esta malformación, describir su cuadro clínico y compararlo con la literatura médica revisada.
MÉTODOS
Se realizó una investigación descriptiva retrospectiva en el Hospital Provincial Docente Clínico Quirúrgico José Ramón López Tabrane, de Matanzas, durante el año 2009, sobre una paciente portadora de la malformación de Arnold Chiari tipo I, quien ingresó en la sala J de dicho centro asistencial, comparándose con una extensa revisión bibliográfica referente a esta entidad.
A la paciente se le solicitó consentimiento informado para la presentación de esta investigación, cuidándose los aspectos básicos de la bioética.
Fuentes de información
El dato primario se obtuvo de la revisión de la historia clínica de la paciente presentada, así como de la entrevista médica realizada por los autores de la investigación.
Procedimiento
La información relativa a esta investigación fue apoyada por un enfoque médico, utilizándose criterios clínicos e imagenológicos en su discusión.
Procesamiento de la información
Los resultados se procesaron en Microsoft Word 2003, empleando una PC Pentium M Celaron 1300 Mhz, con ambiente de Windows XP.
PRESENTACIÓN DEL CASO
Historias clínicas: 519320 y 521215.
Paciente J. R. R, femenina, de 44 años de edad, blanca, de procedencia rural, fumadora consuetudinaria, con antecedentes de enfermedad de Habermann, quien ingresó en la sala J del Hospital Provincial Docente Clínico Quirúrgico José Ramón López Tabrane, de Matanzas, en octubre de 2009, por cuadro de cefalea pulsátil, generalmente hemicraneana izquierda, de intensidad variable, y preferencia matutina, acompañada de trastornos visuales (ambliopía y oscurecimiento visual), vómitos matutinos escasos, en proyectil, parestesias braquiales e inestabilidad en la marcha. Estos episodios se presentan en crisis y recidivas, exacerbándose en los últimos tiempos, pese al tratamiento impuesto con acetazolamida, furosemida y prednisona por neurología desde el año 2004. Además, fue operada de cirugía refractaria en abril de 2009.
Examen físico
Peso habitual: 50 kg
Peso actual: 52 kg
Temperatura: 36.6 ºC
Mucosas: Húmedas y normocoloredas
Piel y faneras: normales.
Respiratorio: murmullo vesicular disminuido en ambos hemotórax, donde se auscultan estertores subcrepitantes aislados. No retracciones intercostales y subcostales, discreto aleteo nasal, no cianosis. Fr: 24/ rpm. Saturación de Oxígeno: 98 %.
Sistema cardiovascular: primer ruido y segundo ruido cardiaco normal, no tercer ruido ni soplo. Los pulsos periféricos de buena amplitud y fuertes. El relleno capilar rápido.
Fc: 188/lpm.
Tensión Arterial: 125/85.
Abdomen: plano, depresible, no distendido, no visceromegalia, no masas palpables.
Sistema nervioso: discreta espasticidad de los brazos y flacidez de las piernas, reflejos osteotendinosos exaltados. Fondo de ojo, papiledema bilateral, disco óptico elevado, bordes borrados, tortuosidad vascular, no pulso venoso. Nistagmo horizontal agotable a la extrema izquierda.
Exámenes realizados
1. Hb: 13 g/l
2. Leucograma: 7,3 x 10/l
3. Plaquetas: 280x10/l
4. Urea: 3,6 mmol/l
5. Creatinina: 82 mmol/l
6. Proteína C reactiva: 2 mg/l
7. Proteínas totales: 77 g/l
8. Albúmina: 34 g/l
9. Bilirrubina total: 28 mmol/l
10. Bilirrubina directa: 8 mmol/l
11. V.S.G: 8 mms
12. Perfil hepático: normal
13. Serologías VIH y VDRL: no reactivas
14. Antígeno de superficie y anticuerpo C: no reactivos
15. A.N.A y ANCA negativos
16. Lipidograma normal
17. Ultrasonido abdominal, tiroideo y ginecológico normales
18. Mamografía: sin alteraciones
19. Panendoscopía y tránsito intestinal sin alteraciones
20. Rx de tórax: negativo
21. L.C.R: Presión de L.C.R: 35 cm de agua, citoquímico, bacteriológico, micológico e inmunológico: normal. Tiene realizada cinco punciones lumbares con valores de presión por encima de 30 cm de agua.
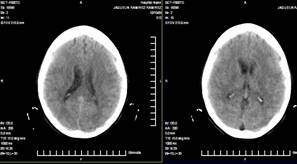
22. TAC de cráneo simple: muestra dilatación de los ventrículos laterales. (Anexo 1)
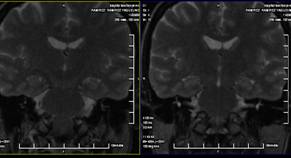
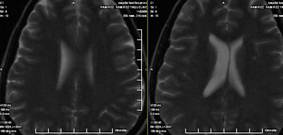
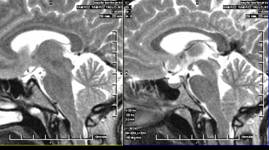
23. RMN de cráneo: Se realiza T2 y T1 sagital, T2 y T1 coronal y T2 axial, observándose silla turca de diámetros normales, con la presencia de un aracnocele intraselar de 9 mm x 5 mm x 8 mm. El quiasma óptico y el tallo hipotálamo hipofisario es normal. Descenso de las amígdalas cerebelosas a través del agujero magno (8 mm), imagen polipoidea isointensa en T2 de 18 mm x 14 mm en el seno maxilar derecho. (Anexos 2 y 3)
24. Campimetría por confrontación: normal
25. Glicemia: 5,18 mmol.
DISCUSIÓN
La hidrocefalia en la paciente se debe a una malformación de Arnold Chiari, en la cual existe un descenso anormal en grado variable de la porción inferior del cerebelo, del bulbo y el cuarto ventrículo hacia el conducto raquídeo a través del agujero occipital (3).
La entidad fue descrita por primera vez en 1891, por Chiari, y en 1894 Arnold describió un caso asociado a mielomeningocele; pero no es hasta 1903 que finalmente Schwalbe y Gredig le otorgaron el nombre de malformación de Arnold Chiari (10).
La hidrocefalia ocasionada por esta malformación se debe al impacto de estas estructuras (cerebelo, tallo y cuarto ventrículo) en el agujero occipital, proporcionando estenosis del acueducto de Silvio y bloqueo del desagüe de LCR del cuarto ventrículo (11).
Ha llamado la atención de muchos autores que puede existir la malformación de Arnold Chiari sin asociación de otras malformaciones (12-20), lo que ha dado lugar a que múltiples teorías que hayan tratado de explicar su aparición sean desechadas, como la de Penfild y Cobor (1938). Esta plantea que la malformación se debía a la tracción sobre el tallo cerebral en la vida embrionaria, a consecuencia de la fijación de la médula espinal a nivel de un meningocele. Otras teorías como la de Peach (1965) y la de Garner (1976), tratan de explicarla a través de una falta en el desarrollo del ángulo protuberancial a causa de una detención precedente, o a través de un estado disráfico.
En revisión bibliográfica realizada (12,13,16-8), se aprecia la asociación frecuente de anomalías óseas en la región cervical como la impresión basilar, platibasia, fusión del atlas y el axis, etc. Esta entidad conduce a la reflexión sobre la correspondencia existente con alteraciones en mayor o menor grado a nivel del cierre del tubo neural, lo que puede justificar su fisiopatogenia.
Lo variado del conjunto de síntomas está en relación con las estructuras afectadas. Cuando existe obstrucción a la circulación del LCR predominan los síntomas de hidrocefalia. En otros casos, los síntomas hacen sospechar de un tumor cerebeloso, una siringomielia, una esclerosis múltiple o diversos tipos de enfermedades degenerativas.
Para realizar su diagnóstico se precisa de las manifestaciones clínicas, así como de los estudios mielográficos cervicales, ya sea por técnica de TC o por RMN. Estas ayudan a identificar perfectamente el diagnóstico así como el tipo de malformación del cual se trata (21,22).
Manifestaciones clínicas
En el tipo II de malformación de Arnold Chiari (con meningomielocele) el problema es esencialmente el de un hidrocéfalo progresivo. Los signos cerebelosos pueden no ser distinguidos en los primeros meses de vida (2). Además, anormalidades de los nervios craneales inferiores pueden presentarse en combinaciones varias. Si el paciente sobrevive hasta el final de la infancia o adolescencia, uno de los síndromes que se presentan con malformación de Arnold Chiari tipo I se pueden presentar (3).
En la malformación de Arnold Chiari tipo I (sin mielomeningocele u otros signos de disrafismo), los síntomas neurológicos pueden no desarrollarse hasta la adolescencia o vida adulta. Los mismos pueden ser (4):
• Incremento de la PIC
• Ataxia cerebelosa progresiva
• Siringomielia (amiotrofia segmental y trastornos sensitivos, con o sin dolor)
Los pacientes pueden presentarse con una combinación de síntomas de los nervios craneales inferiores, cerebelo, bulbo y médula espinal (signos motores y sensitivos de las vías largas), normalmente en conjunción con cefalea (6). A menudo la enfermedad es confundida con esclerosis múltiple o con un tumor del foramen magnun o médula cervical inferior. Los síntomas pueden tener un comienzo agudo después de una extensión, manipulación quiropráctica o extracción dental (3). El hábito físico de tales pacientes puede ser normal, pero cerca del 25 % tiene signos de una hidrocefalia detenida por un cuello corto. Cuando coexisten impresión basilar y ACM es imposible decidir cuál de las dos malformaciones es responsable de los problemas.
Diagnóstico y tratamiento
La Resonancia Magnética Cerebral es el medio de diagnóstico complementario más útil (21-4).
El tratamiento de la impresión basilar y malformación de Arnold Chiari está lejos de ser satisfactoria. Si la progresión es ligera o incierta probablemente es mejor no hacer nada. Si la progresión es cierta y la incapacidad se incrementa, una laminectomía cervical superior y el aumento del foramen magnum está indicado. Con frecuencia este procedimiento para la progresión de la enfermedad resulta en alguna mejoría. El procedimiento quirúrgico debe realizarse con cuidado. La apertura de la duramadre y la manipulación extensa de la malformación puede agravar los síntomas o causar la muerte (20).
Anexo No. 1
Anexo No. 2
Anexo No. 3

REFERENCIAS BIBLIOGRÁFICAS
1. Martí I, García A, Prats JM. Malformación de Chiari tipo I y siringomielia reversible. Neurología. 2003 Mar;18(2):101.
2. López R, Nazar C, Sandoval P, Guerrero I, Mellado P, Lacassie HJ. Malformación de Arnold-Chiari tipo I con siringomielia, trabajo de parto y analgesia neuroaxial. Rev Esp Anestesiol Reanim. 2007 May;54(5):317-21.
3. Plaza M, Barón R, Herraiz P, López L, Aparicio F. Manifestaciones otoneurológicas como presentación de la malformación de Chiari tipo I. An Otorrinolaringol Ibero Am. 2006;33(6):613-22.

