










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev. Med. Electrón. v.33 n.2 Matanzas mar.-abr. 2011
HOSPITAL MILITAR DOCENTE DR. MARIO MUÑOZ MONROY. MATANZAS
Feocromocitoma bilateral: presentación de un caso
Bilateral pheochromocytoma: presentation of a case
AUTORES
Dr. Yuniel Arocha Molina (1)
Dra. Yanet Acosta Piedra (2)
Dra. Blanca Piedra Herrera (3)
E-mail: bcpiedra.mtz@infomed.sld.cu
Dra. Teresa Suárez Díaz (4)
Dra. Ketty Madruga Vázquez (5)
1) Especialista de I Grado en Medicina Interna. Máster en Enfermedades Infecciosas. Profesor Instructor. Hospital Militar Docente Dr. Mario Muñoz Monroy. Matanzas.
2) Especialista de I Grado en Medicina Interna. Profesora Asistente. Hospital Militar Docente Dr. Mario Muñoz Monroy. Matanzas.
3) Especialista de I Grado en Medicina Interna. Profesora Asistente. Hospital Provincial Clínico-Quirúrgico Docente José R. López Tabrane. Matanzas.
4) Especialista de I Grado en Medicina Interna y en Medicina General Integral. Máster en Urgencias Médicas.Hospital Militar Docente Dr. Mario Muñoz Monroy. Matanzas.
5) Especialista de I Grado en Anatomía Patológica. Profesora Instructora. Hospital Militar Docente Dr. Mario Muñoz Monroy. Matanzas.
RESUMEN
El feocromocitoma como causa de hipertensión arterial secundaria tiene una baja incidencia. La edad de presentación más frecuente es entre 30 y 50 años. Puede tener carácter hereditario autosómico dominante. Sus manifestaciones clínicas no dependen de la topografía ni del tamaño del tumor. Puede debutar con complicaciones graves. Se presenta un paciente de 32 años, cuyo padre hipertenso fallece a los 38 años, sin precisar la causa. El paciente ingresa por el debut de una hipertensión paroxística, se demuestra por ultrasonido y tomografía axial computarizada grandes tumores suprarrenales bilaterales, compatibles con feocromocitoma y el paciente fallece cuatro días después del ingreso por un edema agudo del pulmón, en el curso de una emergencia hipertensiva. Comprobándose por anatomía patológica el diagnóstico tanto macro como microscópico, de feocromocitoma.
Palabras clave: feocromocitoma, hipertensión, células cromafines, informes de casos, humanos, masculino, adulto.
SUMMARY
The pheochromocytoma as a cause of secondary arterial hypertension has low incidence. The most frequent age of presentation is between 30 and 50 years old. It can have dominant autosomyc hereditary character. Its clinical manifestation does not depend on the topography or the size of the tumor. It can debut with serious complications. We present the case of a patient aged 32, whose hypertensive father died at the age of 38, without specified cause. The patient entered the hospital for the debut of a paroxysmal hypertension. By means of ultrasound and on-line axial tomography there were found bilateral suprarenal tumors compatible with pheochromocytoma and the patient dies four days after entering the hospital by acute lung edema, during a hypertensive emergency. It was proven, for pathologic anatomy, the diagnosis so much macroscopic as microscopic, of pheochromocytoma.
Key words: pheochromocytoma, hypertension, chromaffin cells, case reports, humans, male, adult.
INTRODUCCIÓN
Los feocromocitomas son tumores del sistema nervioso simpático que se desarrollan a partir de las células cromafines y que se caracterizan por la producción excesiva de catecolaminas. En el 85 % de los casos se localizan en la médula suprarrenal y el 15 % restante son extrasuprarrenales. Se ha estimado que lo presentan alrededor del
Lo infrecuente de esta enfermedad y su forma de presentación en este caso, nos motivó a realizar este trabajo. Es una causa de Hipertensión arterial en pacientes jóvenes, curable, y que en la mayoría de los casos, no se llega al diagnóstico de certeza, lo que da al traste con su vida.
PRESENTACIÓN DEL CASO
Paciente E. A. J. Masculino. Edad: 32 años.
Fecha de ingreso: 23-6-2010
Fecha de egreso: 27-6-2010
Paciente que comienza hace alrededor de un mes con decaimiento marcado asociado a sudoraciones intensas, que ocurren varias veces al día, recuperándose en pocos minutos y al que se constató en el área de salud, en varias ocasiones, cifras elevadas de tensión arterial.
APF: Padre fallecido a los 38 años de edad, no precisa la causa de muerte. Padecía de hipertensión arterial.
Datos positivos al examen físico
TA: 180/120 sólo en una ocasión durante el pase de visita matutino, refería en ese momento intensa cefalea. Se recuperó en 2 horas.
Complementarios realizados
Hb. 14.9 g/L Hto 0.48
Eritrosedimentación: 7mm/h
Leucograma: 4.6x 10 9 /L
Seg: 0.70
Linf: 0.25
Eos:0.03
Glicemia: 5.91 mmol/L
Creatinina: 86.4 MMol/L Ac.
Úrico: 369.5 mmol/L
Colesterol: 6.37 MMol/L
TG: 0.85 MMol/L
ECG: Normal.
Ultrasonido abdominal: Entre el lóbulo derecho y polo superior del riñón derecho se observa una imagen redondeada de contorno bien definido hiperecogénica con áreas de menos ecogenicidad en su interior de 67. 8 X
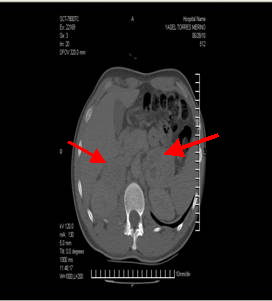
Se indica tomografía axial computarizada (TAC) de abdomen, donde se confirma un proceso expansivo en proyección de la glándula suprarrenal derecha con zonas de necrosis intratumoral que mide 71 x 61 x
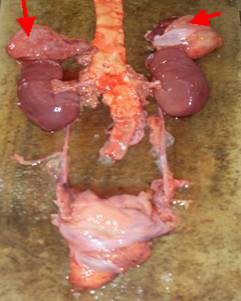
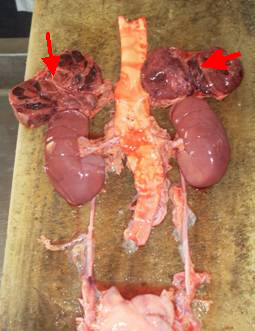
En la necropsia se aprecia tumor bilateral de las glándulas suprarrenales, multilobuladas con áreas de hemorragias que ocupan todo su interior; en el árbol bronquial se aprecia abundante secreción espumosa compatible con un edema agudo del pulmón.
Causa directa de muerte: Edema agudo del pulmón.
Causa intermedia de muerte: Emergencia hipertensiva.
Causa básica de muerte: Tumor bilateral de glándulas suprarrenales
Morfología feocromocitoma |
| Topografía de glándulas suprarrenales (derecha e izquierda) |
|
|
|
|
|
|
|
|
|
Tomografía axial computarizada |
| Foto de la pieza anatómica |
|
|
|
|
|
|
|
|
|
Foto de la pieza anatómica |
| Lámina histológica |

Diagnóstico microscópico: tumor formado por células cromafines con citoplasma finamente granuloso. Compatible con feocromocitoma.
DISCUSIÓN
Este paciente joven ingresa para estudio de una hipertensión arterial de debut, con historia familiar de esta enfermedad, comportándose de forma paroxística y que fallece en el curso de una complicación sobreaguda, sin llegar a imponérsele tratamiento específico, por su corta estadía, al haber ingresado en un estadio avanzado de la enfermedad. No se contó con otros medios diagnósticos, ni hubo tiempo para gestionarlos en otro centro. Esta forma de presentación se refleja en la literatura revisada, donde se plantea que de manera característica, las manifestaciones clínicas son paroxísticas en la gran mayoría de los pacientes. El inicio de las crisis es repentino y el acmé se alcanza a los pocos minutos con una duración total de menos de una hora, pudiendo presentar fallo ventricular izquierdo y edema pulmonar agudo (1,6,7). Nunca se confirmó la causa de muerte del padre, pero llama la atención que fallece a los 38 años y tenía antecedentes de hipertensión arterial. El hecho de tener una topografía bilateral y de presentarse a edad tan temprana, nos hace pensar que tiene un origen hereditario, presentándose en esta enfermedad con carácter autosómico dominante (1,8). La localización más frecuente es derecha (1,9).
La clínica del feocromocitoma puede ser muy variada y no guarda una clara relación con el tamaño, localización o aspecto histológico del tumor (1-3,5,10). En la literatura se plantea que el tamaño más frecuente oscila entre 3 y
No obstante la baja frecuencia diagnóstica del feocromocitoma, es una enfermedad a tener en cuenta en todos los pacientes jóvenes que ingresan por hipertensión arterial, y debe dársele importancia a todos los datos clínicos, además de la historia familiar, con el objetivo de hacer su diagnóstico precoz, ya que es una entidad curable.
REFERENCIAS BIBLIOGRÁFICAS
1. Landsberg L, Young JB. Pheochromocytoma. En: Harrison 's Principles of Internal Medicine. 17 th ed, New York: Mc Grw Hill Elsevier; 2007.
2. Herrera RN et al. Feocromocitoma asociado a neurofibromatosis de Von Recklinghausen. Medicina. 2007 Sep-Oct;(67)5.
3. Touiti D, Seket B, Deligne E. Bilateral adrenal pheochromocytomas in Von Hippel-Lindau disease. Ann Urol. 2001;35:323-8.
4. Caruso G. Feocromocitoma con catecolaminas reiteradamente normales. Rev Argent Cardiol. 1999;(67):805-7.
5. Mateo A, Rentería M. Feocromocitoma: revisión o manejo quirúrgico. Rev Hosp Gral Dr. M Gea González. 2000;3(4):170-81.
6. Cabrera Gámez M. Feocromocitoma. Presentación de un caso clínico. Rev Cubana Endocrinol. 2008 Mayo-Ago;19(2).
7. Manger WM, Gifford RW Jr. Pheochromocytoma: a clinical overview In: Laragh JH, Brenner BM, eds. Hypertension: Pathophysiology, Diagnosis and Management. 2nd ed. New York: Raven Press Ltd; 1995. p. 2225-44.
8. Amberson JB, Vaughan ED, Gray GF. Flow cytometric determination of nuclear DNA content in benign adrenal pheochromocytomas. Urology. 1987;30(2):102-4.
9. Knop G L. Feocromocitoma cardíaco Rev Argent Cardiol. 2006 Sep-Oct;74(5).
10. Szolar D, Korobkin M, Reittner Pia, Berghold A, Bauernhofer T, Trummer H, et al. Adrenocortical Carcinomas and Adrenal Pheochromocytomas: Mass and Enhancement Loss Evaluation at Delayed Contrast-enhanced CT. Radiology [seriada en
CÓMO CITAR ESTE ARTÍCULO
Arocha Molina Y, Acosta Piedra Y, Piedra Herrera B, Suárez Díaz T, Madruga Vázquez K. Feocromocitoma bilateral. Presentación de un caso. Rev méd electrón [Seriada en línea] 2011;33(2). Disponible en URL: http://www.revmatanzas.sld.cu/revista%20medica/ano%202011/vol2%202011/tema17.htm [consulta: fecha de acceso]

