










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev. Med. Electron. vol.37 no.1 Matanzas ene.-feb. 2015
ARTÍCULO ORIGINAL
Reacciones adversas en ensayos clínicos con nuevos fármacos conducidos en Brasil. Años 2000 y 2012
Adverse reactions in clinical trials carried out in Brazil with new drugs. Years 2000-2012
DrC. Maria Rita Garbi Novaes,I Dra. Elsa Pastor,II Dra. Alice Garbi Novaes,III DrC. Dirce Guilhem,IV DrC. Roberto Cañete VillafrancaV
I Universidad de Brasilia. Brasilia, Brasil.
II Secretaria de Salud del Distrito Federal. Brasilia, Brasil.
III Universidad Católica de Brasilia. Brasilia, Brasil.
IV Facultad de Ciencias Sociales de la Universidad de Brasilia. Brasilia, Brasil.
V Centro Provincial de Higiene, Epidemiología y Microbiología. Matanzas, Cuba.
RESUMEN
Introducción: los ensayos clínicos constituyen un diseño de tipo experimental utilizado para evaluar cualquier actuación diagnóstica y terapéutica aplicada a humanos.
Objetivo: evaluar los relatos de reacciones adversas a medicamentos, en cuanto a causalidad, gravedad, edad y grupos vulnerables en protocolos de ensayos clínicos con nuevos fármacos analizados en el Comité de Ética de la Investigación de la Secretaria de Salud de Brasilia. Distrito Federal, Brasil.
Método: estudio observacional, descriptivo y transversal, en la modalidad de estudio de caso, se consideró como universo de trabajo los proyectos evaluados por el Comité de Ética en Investigación-CEI/SES/DF en el período comprendido entre los años 2000 y 2012.
Resultados: el 59 % de las investigaciones se correspondieron con estudios nacionales y el 41 % con estudios internacionales. La industria farmacéutica fue la fuente financiadora en el 65 % de los casos y en el 41 % de ellos existió participación extranjera. De los medicamentos estudiados el 19 % no poseían registro en la Agencia Nacional de Vigilancia Sanitaria-ANVISA y solo en el 9,5 % de los ensayos se utilizó grupo control. Las reacciones adversas fueron consideradas como relacionadas a los medicamentos, posibles o improbables en el 15,7 %, 19,8 % y 63,6 %, respectivamente.
Conclusiones: la mayor parte de las investigaciones en las que se evaluaron nuevos medicamentos fueron conducidas por instituciones brasileñas y en la mayoría se recibió financiación internacional. La falta de registro de algunos de los nuevos productos en la agencia nacional reguladora y la no existencia de grupo control, en diferentes ensayos clínicos, fueron limitaciones identificadas en el proceso de evaluación. Se identificó la no existencia de modelos únicos legalmente validados para la evaluación de los eventos adversos a los medicamentos.
Palabras clave: ética en investigación, ensayos clínicos, nuevos medicamentos.
ABSTRACT
Background: clinical trials are an experimental kind design used to assess any diagnostic and therapeutic performance applied to human beings.
Aim: to assess the reports of adverse reactions to drugs, according to causality, seriousness, age and vulnerable groups in protocols of new drugs clinical trials analyzed in the Research Ethic Committee of Brasilia Health Secretariat, Federal District, Brazil.
Method: cross-sectional, descriptive, observational study of case study kind. As universe of work, we took into account the projects assessed by the Research Ethic Committee of Brasilia Health Secretariat, Federal District (CEI/SES/DF in Portuguese) in the period from 2000 to 2012.
Outcomes: 59 % of the research corresponded to national studies and 41 % to international ones. The pharmaceutical industry was the financeable source in 65 % of the cases and there it was foreign participation in 41 % of them. 19 % of the studied drugs were not registered in the National Agency of Sanitary Surveillance (ANVISA in Portuguese) and control group was used in only 9.5 % of the trials. The adverse reactions were considered as related to drugs, possible or improbable in 15.7 %, 19.8 % and 63.6 %, respectively.
Conclusions: most of the research where new drugs were assessed was carried out by Brazilian institutions and most of them received international financial support. The lack of registration of several new products in the national regulatory agency and not using control groups were limitations identified during the assessing process in several clinical trials. We identified the inexistence of unique models legally validated for the evaluation of adverse events to drugs
Key words: ethics in research, clinical trials, new drugs.
INTRODUCCIÓN
La investigación clínica tiene entre sus objetivos analizar y evaluar la investigación con seres humanos en las áreas biomédicas, tanto para adquirir nuevos conocimientos o con fines terapéuticos, en la búsqueda para elevar la calidad de vida de la población.(1,2)
Los ensayos clínicos constituyen un diseño experimental que permiten bajo evidencia sólida evaluar cualquier nueva actuación diagnóstica y terapéutica en humanos. La finalidad del ensayo clínico es constatar la eficacia de una intervención médica en condiciones ideales, a menudo, alejadas del medio asistencial. La investigación es costosa en recursos económicos, humanos y organizativos, especialmente cuando se trata de estudios multicéntricos.(1,3)
Los resultados de esos estudios experimentales deben conducir a progresos en la salud y el bienestar de la comunidad, además de generar conocimientos. Es claro que ese empeño no se alcanza sin conflictos y dilemas pues deberá ser razonable defender el derecho y la libertad de investigar siempre y cuando se respete la dignidad humana. La fragilidad y el sufrimiento del enfermo deben recibir cuidado especial y la eventual cura debería estar disponible para los individuos una vez terminado el estudio, evitándose con ello la explotación.(1)
La investigación clínica realizada para el desarrollo de nuevos fármacos es un proceso largo y costoso (promedio de 10 a 12 años con un costo de 300 billones de dólares). Gran parte de las investigaciones clínicas se destinan a realizar estudios comparativos complejos que exigen la aleatorización de los participantes y para ello se exige que los protocolos tengan validez científica, respeten los derechos de los pacientes y sean éticamente correctos.(3)
Los ensayos clínicos con fármacos deben contener aspectos metodológicos en los que queden claramente expuestos los nombres de los medicamentos a utilizar, así como las dosis en que serán ofrecidos (tanto en grupos estudio como en grupos control). De igual forma se deben definir claramente las vías de administración, el posible uso de fármacos concomitantes, así como la forma en que serán evaluadas la eficacia y la seguridad. Especial atención se debe ofrecer a las medidas para la monitorización de los efectos adversos y la comunicación, de todo lo relacionado con el proceso, al paciente, las agencias sanitarias, regulatorias, el patrocinador y los comités de ética de la investigación.
Los estudios necesitan ser conducidos utilizando buenas prácticas clínicas que no son más que un conjunto de condiciones que debe cumplir las investigaciones de este tipo para garantizar la validez de los datos y resultados obtenidos así como su reproducibilidad.(1-6)
En la fase pre clínica se desarrollan estudios “in vitro” en los que se comprueba la actividad farmacológica y la seguridad del producto. Generalmente, en esa fase, el 90 % de las sustancias bajo investigación son desechadas. Si la actividad farmacológica es adecuada y el perfil de toxicidad seguro, se continúa con las diferentes fases de los ensayos clínicos controlados incluyendo la fase de post- comercialización. Todas las etapas de le investigación deben contar con la aprobación en un comité de ética de la investigación.(3,7)
La fase clínica I, con una duración aproximada de 5 a 7 años, se conduce en voluntarios con buena salud física y mental y generalmente en número pequeño. El objetivo de esta fase es evaluar la seguridad en seres humanos.(3,7) Las fase clínicas II, III con duración de 3 a 7 años, tienen como objetivo evaluar la efectividad y los efectos indeseables de los medicamentos. Consideran una casuística mayor y generalmente se procuran estudios en los que los diseños sean a doble ciego, controlado y multicéntricos.(6,7)
En la fase clínica IV, con duración de cerca de 3 a 5 años, ocurre la aprobación, registro del medicamento en las agencias regulatorias locales e internacionales, comercialización y la divulgación a través de estrategias diversificadas de marketing.(3,7)
El registro de un medicamento por las agencias regulatorias nacionales e internacionales como la agencia Norteamericana Food and Drug Administration (FDA) autoriza el empleo de los medicamentos para su comercialización, lo que incluye la protección de los derechos de la población en cuanto a calidad, seguridad y eficacia de los medicamentos para la enfermedad específica.(6)
La selección y el reclutamiento de los sujetos deben garantizar la reducción al mínimo los riesgos para los mismos, a la vez, que se maximizan los beneficios sociales y científicos de los resultados de la investigación. La selección equitativa de sujetos se justifica por el principio de la equidad distributiva.(7) Sin embargo, en ensayos clínicos donde se testan los efectos de los fármacos, los pacientes pueden tener reacciones adversas. Esas reacciones son clasificadas, en cuanto a su gravedad, en leves, medianas o graves y en cuanto a causalidad, como relacionada o no relacionada al fármaco en estudio.(8)
Conceptualmente la reacción adversa a un medicamento es cualquier efecto perjudicial o indeseable no intencional que aparezca, luego de la administración de un medicamento en dosis normalmente utilizadas en humanos, para la profilaxis el diagnóstico o el tratamiento de una enfermedad. Siendo así, no son consideradas reacciones adversas a medicamentos o efectos adversos que aparecen al utilizar dosis mayores de las habituales.(8) Los relatos de reacciones adversas a los fármacos son importantes para conocer la incidencia de las mismas y para detallar las características de esos eventos.
La decisión de los individuos de participar en ensayos clínicos controlados debe ser libre. Los sujetos deben recibir información en cantidad y calidad suficientes sobre la finalidad de la investigación, las características de la enfermedad que padecen, el pronóstico de la entidad, el tipo de estudio en el que serán incluidos y las molestias de los procedimientos a los que serán sometidos, así como los riesgos, los beneficios y las alternativas a la investigación, permitiéndoles cambios de opinión para retirarse, sin sanción o coerción del estudio, en el que sean incluidos. El sujeto debe entender su situación clínica y su decisión debe ser autónoma.(4,7,8)
Antes de iniciarse, toda investigación en seres humanos, debe ser revisada por un comité de ética cuya responsabilidad es asegurar la preservación de la seguridad, los derechos y el bienestar de los participantes en la investigación.
Las investigaciones clínicas están normalizadas en Brasil a través de la resolución 196/96 del Consejo Nacional de Salud del Ministerio de Salud. Esta resolución dispone las directrices que rigen la investigación con seres humanos y crea un sistema nacional de validación ética de la investigación representada por el Consejo Nacional de Ética de la Investigación (CONEP) y los diferentes Comités de Ética de la Investigación disgregados por todo el territorio nacional. Es importante mencionar que el Tratado del Mercado Común del Sur (MERCOSUR) elaboró la Resolución No 129/96 donde se disponen las buenas prácticas clínicas.(7-10)
Todas las investigaciones desarrolladas en seres humanos en Brasil deben ser evaluadas por un Comité de Ética de la Investigación (CEI) que considere la reglamentación del Ministerio de la Salud Brasileño (Resolución CNS 196/96, Brasil) y con la No 39 de 05/06/2008 de la Agencia Nacional de Vigilancia Sanitaria (ANVISA) y con el Manual de Buenas Prácticas Clínicas elaborada por la Organización Panamericana de la Salud en 2005.(7-9) Brasil tiene alrededor de 600 CEI certificados.
Con el rápido desarrollo científico-tecnológico la reflexión ética sobre los procesos de validación de los proyectos debe seguir siempre las directrices contenidas en la Resolución 196/96 y en la Resolución actual 466/12. Las resoluciones publicadas por el Consejo Nacional de Salud del Ministerio de Salud de Brasil deben también ser utilizadas en el proceso de validación ética de los protocolos de investigación clínica para dar respuesta a los nuevos desafíos por los que transita la ciencia en el país y se exponen a continuación :
Resoluciones publicadas por el Consejo Nacional de Salud, Ministerio de Salud, Brasil, utilizadas en el proceso de validación ética de los protocolos de investigación clínica.(7,8)
- Resolución CNS 466/12 del año 2012: Directrices y normas que regulan la investigación en seres humanos.
- Resolución nº 240/97 del año 1997: Define la representación de los usuarios por los Comité de Ética de la Investigación.
- Resolución CNS 251/97 del año 1997: Norma complementaria para el área temática especial de nuevos medicamentos, vacunas y medios diagnósticos. Delega en el Comité de Ética de la Investigación el análisis de los proyectos en esa área, cuando no fueron enmarcados en otras áreas especiales.
- Resolución CNS 292/99 del año 1999: Normas específicas para la aprobación de protocolos de investigación con cooperación internacional, manteniendo el requisito de aprobación por la Comisión Nacional de Ética de la Investigación, posterior a la aprobación del Comité de Ética de la Investigación local.
- Resolución CNS 303/00 del año 2000: Norma complementaria para el área de reproducción humana, estableciendo sub-áreas que deben ser analizadas por la CONEI y delegando en los CEI el análisis de otros proyectos de esa área temática.
- Resolución CNS 304/00 del año 2000: Norma complementaria para el área de Investigaciones con poblaciones indígenas.
- Resolución CNS 340/04 del año 2004: Directrices para análisis ético y tramitación de los proyectos de investigación en el área temática especial de Genética humana.
- Resolución CNS 346/05 del año 2005: Proyectos multicéntricos.
- Resolución CNS 347/05 del año 2005: Directrices para análisis ético de proyectos de investigación que involucren el almacenamiento de materiales o la utilización de materiales de investigaciones anteriores.
- Resolución CNS 370/07 del año 2007: Registro y acreditación de los CEI en la CONEI y renovaciones de registro de CEP.
El CEI de la Secretaria de Salud del Distrito Federal (SES/DF) se crea el 18 de junio de 1997 para defender los intereses, la dignidad y la integridad de los participantes de los ensayos clínicos e investigaciones en seres humanos que se desarrollasen en los 18 hospitales y 89 unidades de atención básica de salud adscriptos a la institución. El proceso de validación de la calidad de las actividades complementarias desarrolladas en una institución de salud, constituye también, requisito importante para garantizar la transparencia del servicio e identificar posibles debilidades que permitan proponer cambios y definir nuevas metas de forma que se garantice la seguridad de los participantes en las investigaciones.
El objetivo de este estudio es evaluar los relatos de reacciones adversas a medicamentos, en cuanto a relación de causalidad, gravedad, edad y grupos vulnerables en protocolos de ensayos clínicos fase III y IV con nuevos fármacos sometidos al Comité de Ética de la Investigación de la Secretaria de Salud de Brasilia, Distrito Federal, Brasil.
MÉTODOS
Se desarrolló un estudio observacional, descriptivo, de corte transversal, en la modalidad de estudio de caso. Los datos fueron colectados a partir de la información contenida en las copias de proyectos archivados en el CEI/SES/DF en el periodo de tiempo comprendido entre 2000 y 2012. Los datos fueron almacenados y procesados utilizando el programa Microsoft- Excel, versión 2007.
Para el análisis de las reacciones adversas a los medicamentos (causalidad y gravedad) fueron incluidas los proyectos de farmacología clínica aprobados y conducidos en los hospitales, unidades básicas de salud y otros servicios ofrecidos por la SES/DF. Los proyectos de farmacología clínica fueron seleccionados por el título y el número de identificación con los que fueron archivados en el CEI/SES/DF. Las informaciones fueron colectadas de las hojas de presentación de los proyectos, del cuerpo de esos documentos y de las notificaciones de reacciones adversas a los fármacos.
Las hojas de presentación acompañaron los proyectos de investigación y constituyeron documentos fundamentales para la legalización e inserción de los proyectos en el sistema CEI/CONEI del Ministerio de Salud de Brasil. Las informaciones en ellas, reflejadas fueron esenciales para la conformación de los bancos de datos relacionados con la tramitación de proyectos en todo el país y son automáticamente incluidas en el Sistema Nacional de Informaciones sobre investigaciones que involucran seres humanos.(11) En esas hojas deben estar incluidos todos los datos relacionados con la investigación incluyendo los datos del investigador principal, el director de la institución donde se ejecutará el trabajo, quienes serán los participantes del estudio, las posibles fuentes de financiamiento, etc.(11)
Las reacciones adversas a los medicamentos fueron obtenidas de los ensayos clínicos conducidos durante el año 2009 y clasificadas en cuanto a causalidad, gravedad de acuerdo con parámetros establecidos por la Organización Mundial de la Salud (OMS).
Los resultados fueron codificados y analizados mediante técnicas de estadística descriptiva y frecuencia relativa. El protocolo de trabajo de este estudio fue evaluado y aprobado por el Comité de Ética de la Investigación de la SES/DF bajo el Código nº155/09.
RESULTADOS
En el periodo de tiempo comprendido entre 2000 y 2012 fueron evaluados por el CEI-SES-DF 2021 proyectos de investigación procedentes de diferentes instituciones de salud pertenecientes o no a la Secretaria de Salud del Distrito Federal (Gráf.1).
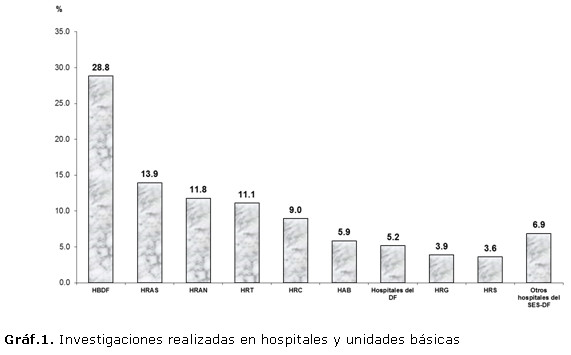
Luego del análisis pertinente el 92 % de los proyectos presentados fueron aprobados. Sin embargo, otro 3 % fue rechazado y el 5 % restante retirados. Una vez recibida la notificación de aceptación emitida por el CEI/SES/DF, los proyectos correspondientes con ensayos clínicos controlados que evaluaban nuevos medicamentos fueron enviados para una revisión adicional por parte del CONEP. De esos proyectos el 4,8 % no fueron aprobados por la instancia superior, 14,3 % no fueron conducidos en el DF y 22 % fueron suspendidos o no se concluyeron. Solamente el 9,5 % de los investigadores principales de esos estudios envió la relatoría final al CEI/SES/DF.
El reducido número de proyectos no aprobados es consecuencia de las acciones educativas promovidas por el comité. Una vez que el proyecto es evaluado por el relator y discutido en las reuniones del CEI/SES/DF, el investigador principal del estudio es notificado sobre la decisión del órgano. Si el parecer fuera desfavorable, se solicita por escrito la aclaración de las limitaciones encontradas. En ese caso la oficina de coordinación del comité emite las orientaciones pertinentes para que el proyecto sea sometido nuevamente al proceso de validación ética.
El gráfico 2 muestra la distribución de investigaciones clínicas con nuevos medicamentos por patrocinadores. CEI/SES/DF, 2000- 2012. La mayoría de ellas, 57,7 %, fueron financiadas por laboratorios farmacéuticos.

La mayoría de las investigaciones (82 %) fueron conducidas en unidades propias de la SES-DF, de ellas el 58 % en unidades hospitalarias y el 17 % en unidades básicas de salud.
De los 2021 proyectos clínicos evaluados por el CEI/SES/DF en el periodo de 2000-2012, el 35 % se correspondieron con farmacología clínica. Los ensayos clínicos controlados correspondientes al área de farmacología clínica conducidos durante el año 2009 fueron incluidos en el análisis de las reacciones adversas a los medicamentos.
El análisis de esos estudios mostró que de los 63 proyectos evaluados en ese periodo, el 59 % se correspondían con estudios nacionales y el resto, 49 %, con investigaciones internacionales. La industria farmacéutica financió 65 % de los estudios y en el 46 % de ellas, hubo participación internacional.
El 19 % de los medicamentos utilizados en los diferentes ensayos clínicos no poseían registro en la Agencia Nacional de Vigilancia Sanitaria (ANVISA) y solo el 9,5 % de los estudios utilizó grupo control.
Las notificaciones de reacciones adversas a los medicamentos fue realizada a partir de formularios validados por CIOMS, ANVISA o por modelos propios desarrollados por las industrias farmacéuticas patrocinadoras de los estudios. No existen en la actualidad modelos únicos legalmente validados en Brasil para la evaluación de los eventos adversos a los medicamentos.
Las reacciones adversas a los medicamentos fueron clasificadas en relación con edad, causalidad (definida, probable, posible, improbable, condicionada e inaccesible) y en cuanto a gravedad (gráficos 3 y 4). Considerando la gravedad, las reacciones adversas fueron clasificadas como: letales (22 %), graves (38 %), moderadas (29 %) y leves (11 %). Las reacciones adversas a los medicamentos fueron consideradas por los investigadores como con causalidad probable en relación con el medicamento utilizado en el 15,7 % de los casos, posible en el 19,8 %, e improbable en el 63,6 %. Los investigadores consideraron que el 40 % de las reacciones adversas letales y el 35 % de las graves se correspondieron con causalidad probable y posible respectivamente.
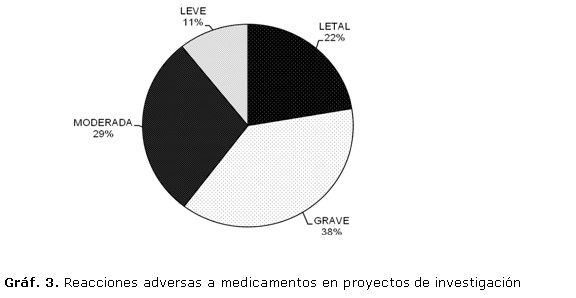
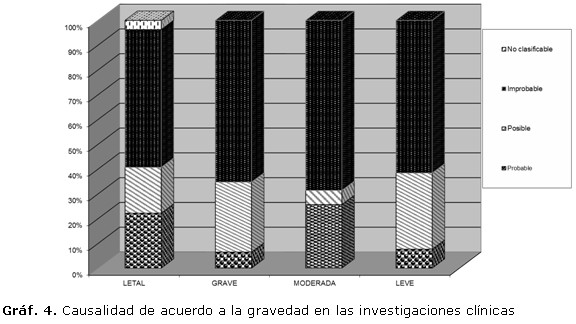
Considerando las edades de los participantes en estudios con nuevos medicamentos, no fue observado un predominio en edades pediátricas o en grupos vulnerables (gráfico 5). Los grupos vulnerables tienen un respaldo legal a partir de resoluciones específicas que rigen el funcionamiento de los CEI y los investigadores (Resolución 466/12 del Consejo Nacional de Salud- Ministerio de Salud, Brasil.

DISCUSIÓN
El funcionamiento articulado de los CEI en Brasil se encuentra limitado en la actualidad, en lo referente al manejo de las reacciones adversas a medicamentos. Eso determina que su función quede reducida a tomar nota de la ocurrencia de esas reacciones, a partir de la información que ofrecen los investigadores principales de los estudios pero sin la existencia clara, en muchas ocasiones, de los procesos que fueron ejecutados para minimizar la ocurrencia de las reacciones y complicaciones asociadas. Los CEI deben, adicionalmente, remitir la información colectada a los cuerpos competentes como la Agencia Nacional de Vigilancia Sanitaria (ANVISA) y CONEP para que sean tomadas las medidas administrativas que procedan de acuerdo a su competencia.
En el caso de los proyectos de farmacología clínica para el desarrollo de nuevos medicamentos (Fases I a III), luego de la notificación de las reacciones adversas a los medicamentos, la información relacionada debe ser enviada a los CEI pertinentes, los patrocinadores, la dirección del centro que funge como sitio de investigación y en los estudios de Fase IV a las oficinas de farmacovigilancia. Esas instancias, en el caso de Brasil, se supeditan a ANVISA y tienen un flujo operacional que debe ser ejecutado en esos casos.
La CEI/SES/DF ha recibido notificaciones de diferentes investigadores responsables de investigaciones clínicas en las que se ha evidenciado alteraciones de los proyectos inicialmente aprobados. Dentro de esas alteraciones las más comunes son: las variaciones en las dosis administradas a diferentes sujetos y la ausencia de exámenes de laboratorio o variaciones en su frecuencia de acuerdo al diseño original.
Esas desviaciones pueden ocurrir tanto en Brasil como en otros países, fundamentalmente cuando el estudio es multicéntrico. En esos casos el CEI/SES/DF ha cuestionado a los investigadores, fundamentalmente, por el posible efecto dañino hacia los sujetos en investigación.
Las actividades educativas conducidas durante 10 años por parte del CEI/SES/DF han determinado, por un lado la mejora en la calidad de lo que se hace y por otro, la concientización de los investigadores, así como la mejora de los proyectos que someten a consideración de la instancia. Ambos resultados han fortalecido la posición del fórum y su legitimización como entidad que salvaguarda la seguridad de los sujetos bajo investigación. Sin embargo, parte del proceso queda aún en manos de investigadores, por lo que la labor educativa y el acompañamiento de los proyectos debe ser una tarea continua.
La evaluación realizada en este estudio fue posible gracias a la ardua labor de preservación de la documentación realizada por los representantes del CEI/SES/DF desde su creación. La información relacionada con las investigaciones conducidas nunca debe ser desechada debido a que son las pruebas de lo realizado y permiten, en última instancia, identificar la ruta crítica de la información y detectar ilegalidades.
La necesidad de un adecuado acompañamiento de las investigaciones, así como del análisis de los eventos adversos notificados en los ensayos clínicos controlados deberían ser elementos de prioridad para los diferentes sistemas de vigilancia sanitaria, en especial, los Comités de Ética en Investigación.
Este estudio evidencia que la mayor parte de las investigaciones en las que se evaluaron nuevos medicamentos fueron conducidas por instituciones brasileñas y en gran parte de ellos se recibió financiación internacional. La falta de registro de algunos de los nuevos productos en la agencia nacional reguladora y la no existencia de grupo control en diferentes ensayos clínicos, fueron limitaciones identificadas en el proceso de evaluación de nuevos fármacos en la Secretaria de Salud de Brasilia. Se identifica la no existencia de modelos únicos legalmente validados en Brasil para la evaluación de los eventos adversos a los medicamentos.
REFERENCIAS BIBLIOGRÁFICAS
1- Cohen MG. Latinoamérica en los ensayos clínicos internacionales: ¿Donde está la diferencia? ¿Son los pacientes, los médicos o el sistema? Rev Argent de Cardio. 2003;71(1):6-15. Citado en LILACS; 354454.
2- Anderson EE, DuBois JM. The need for evidence-based research ethics: A review of the substance abuse literature. Drug and Alcohol Dependence. 2007;86:95-105. Citado en PubMed; PMID:1693085.
3- Guilhem D, Novaes MRCG. Ética e Pesquisa Social em Saúde. In: Fleischer, Schuch P. Ética e Regulamentação na Pesquisa Antropológica. Brasilia : Letras Livres Ed UnB; 2010 . p.248.
4- Dal-Ré Saavedra R, Casas A, de los Reyes López M, Gomis R, Avendaño C, Gracia Guillén DM, Moreno González A, et al. Comités éticos de investigación clínica y ‘’dictamen único’’ de los ensayos clínicos multicéntricos. Med Clin [Internet]. 2003 [citado 3 Abr 2011];120(5):180-88. Disponible en http://dialnet.unirioja.es/servlet/articulo?codigo=307523
5- Moher D, Hopewell, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 Explanation and Elaboration: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c869. Citado en PUBMED; PMID: 20332511.
6- Garbi Novaes MR, Lolas F, Quezada A. Ética y Farmácia. Una Perspectiva Latinoamericana. Monografias de Acta Bioethica n° 02. Programa de Bioética da OPS/OMS. Washington, D.C: OPS; 2009.
7- Garbi Novaes MR, Guilhem D, Lolas F. Dez años de Experiencia do Comitê de Ética em Pesquisa da Secretaria de Saúde do Distrito Federal, Brasil. Acta Bioéth [Internet]. 2008 [citado 3 Abr 2011];14(2):185-92. Disponible en: http://www.scielo.cl/scielo.php?pid=S1726-569X2008000200008&script=sci_arttext&tlng=e
8- Novaes MRCG, Guilhem D, Lolas F. Ethical Conduct in Research Involving Human Beings In Brazil.Arq Med [internet]. 2009 [citado 3 Abr 2011];23(4):145-50. Disponible en: http://www.scielo.oces.mctes.pt/pdf/am/v23n4/v23n4a02.pdf
9- Mercosul. Grupo Mercado Comum. Resolução nº 129/96. Boas práticas clínicas [internet]. 1996 [citado 3 Abr 2011]. Disponible en: http://www.ufrgs.br/bioetica/bpcmerco.htm
10- Ministério da Saúde. Conselho Nacional de Saúde. Sistema nacional de informações sobre ética em pesquisas envolvendo seres humanos–SISNEP. Brasília: CNS/ ;2004 [citado 3 Abr 2011]. Disponible en: http://conselho.saude.gov.br/Web_comissoes/conep/aquivos/sisnep/apresentacao.ppt
11- Glickman SW, Mchutchison JG, Peterson ED, Cairns ChB, Harrington RA, Califf RM, et al. Ethical and Scientific Implications of the Globalization of Clinical Research. The New England Journal of Medicine [Internet]. 2009 [citado 3 Abr 2011];360(8):816-23.Disponible en: http://www.nejm.org/doi/full/10.1056/NEJMsb0803929
Recibido: 7 de noviembre del 2014.
Aceptado: 15 de diciembre del 2014.
Maria Rita Garbi Novaes. Coordenação do Comitê de Ética em Pesquisa- FEPECS/SES-DF. SHIS-QI-09- conj. 06- casa 14 - Lago Sul- Cep: 71.625.060. Correo electrónico:ritanovaes@ig.com.br
