










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev.Med.Electrón. vol.39 no.1 Matanzas ene.-feb. 2017
PRESENTACIÓN DE CASOS
Hemocromatosis herediataria Tipo I. Presentación de un caso
Type I hereditary hemocrhomatosis. Case presentation
Dra. Sahilí Corrales Alonso, Dra. Mariuska Morales Díaz, Dra. Marlenin Estévez López, Dr. Abel Luis Díaz Borroto, Dra. Nereida Del Pilar Álvarez Vega, Dr. Homero Celestrín Tápanes
Hospital Universitario Clínico Quirúrgico Comandante Faustino Pérez Hernández. Matanzas, Cuba.
RESUMEN
La hemocromatosis hereditaria es una enfermedad genética de difícil diagnóstico, en estadios iniciales, ocasionada por alteraciones en el metabolismo del hierro; que conllevan a su depósito en diversos tejidos y como resultado una gran morbilidad en los pacientes afectados. A través de este trabajo se realizó la presentación del primer caso diagnosticado por gastroenterólogos, en el Hospital Faustino Pérez de Matanzas. El paciente debutó con síntomas relacionados con la esfera endocrina como: impotencia, pérdida de la líbido y de la eyaculación. Después de efectuar los estudios correspondientes se concluyó como un hipogonadismo hipogonadotrópico post puberal, por presentar elevación de las enzimas hepáticas. Fue remitido a consulta de Hepatología donde se completó su estudio, confirmándose el diagnóstico de hemocromatosis hereditaria tipo I, a través de biopsia hepática y estudios genéticos.
Palabras clave: hemocromatosis hereditaria, cirrosis por depósito de hierro, sobrecarga de hierro.
ABSTRACT
Hereditary hemocrhomatosis is a genetic disease of difficult diagnosis in its early stages, caused by alterations in the iron metabolism; it leads to iron storage in several tissues and consequently to a great morbidity in affected patients. In this work, we presented the first case diagnosed by gastroenterologists in the Hospital Faustino Pérez, of Matanzas. The patient began with symptoms related with the endocrine sphere like impotence, and lost of libido and ejaculation. After finishing the correspondent studies, we arrived to the conclusion of post-pubertal hypogonadotropic hypogonadism: the patient presented hepatic enzymes elevation. He was referred to Hepatology consultation where the study was finished, confirming the diagnosis of Type I hereditary hemocrhomatosis through biopsy and genetic studies.
Key words: hereditary hemocrhomatosis, cirrhosis by iron storage, iron overload.
INTRODUCCIÓN
La hemocromatosis hereditaria (HH) o primaria incluye una variedad de síndromes crónicos de origen genético que resultan de un error congénito en el metabolismo del hierro, en el que un aumento de su absorción intestinal, causa la sobrecarga y el depósito progresivo en las células parenquimatosas de tejidos pertenecientes a diversos órganos; provocando un deterioro estructural y funcional de los mismos y por consecuencia la aparición de múltiples complicaciones en los pacientes que la padecen.(1-4)
La enfermedad fue descubierta por Armand Trousseau, en 1865, quien describió un síndrome clínico representado por diabetes, piel bronceada y cirrosis hepática, pero no fue hasta 1889 que se reconoció mundialmente con el nombre de Hemocromatosis. En 1996 se identificaron el gen HFE y las dos principales mutaciones asociadas a esta enfermedad (C282Y y H63D), sin embargo en el año 2000 se demostró que un grupo de pacientes no presentaban las alteraciones genéticas descritas cuatro años antes, ni ninguna otra asociada al gen HFE. En el año 2004 se identificaron nuevas mutaciones en otro grupo de genes, todos implicados en la codificación de diferentes proteínas que intervienen en alguna de las etapas del metabolismo del hierro, esto permitió actualizar la clasificación de la HH, quedando establecidas cinco variantes clínico- genéticas: la tipo I, II-A, II-B, III y IV.(1,5,6)
Con el presente artículo los autores se proponen presentar el primer caso de hemocromatosis hereditaria tipo I diagnosticado por especialistas del servicio de Gastroenterología del Hospital Faustino Pérez Hernández de Matanzas.
PRESENTACIÓN DEL CASO
Paciente ECG, masculino, de 28 años de edad, raza mestiza, procedencia rural, con antecedentes de salud anterior, que acude a consulta de Endocrinología del Hospital Faustino Pérez Hernández, por notar disminución del tamaño de sus testículos, "dificultades en la erección" acompañado de pérdida de la eyaculación y de la líbido, desde hacía aproximadamente un año. Comenzó a estudiarse realizándose los correspondientes estudios hormonales (FSH, LH y Testosterona disminuidos), el caso se concluyó como un hipogonadismo hipogonadotrópico post puberal. Se impuso tratamiento con testosterona y gonodatropinacoriónica con discreta mejoría de sus síntomas. (Fig. 1)

Se decidió entonces continuar el estudio con el objetivo de encausar dicho trastorno endocrino, por lo que se realizaron otros complementarios, en los que se detectó un aumento de los valores de las enzimas hepáticas, motivo por el cual fue remitido al servicio de Gastroenterología de dicho centro.
Se recibió al paciente en Consulta Provincial de Hepatología no obteniéndose del interrogatorio datos significativos como: antecedentes patológicos personales, ni familiares de enfermedad hepática previa, así como tampoco síntomas relacionados con enfermedad hepática pasada o actual.
Se reportó por parte del paciente "decaimiento y fatigas" a los esfuerzos físicos, al examen físico se detectó en ese momento los siguientes hallazgos de significación clínica: mucosas: con discreto tinte ictérico en esclerótidas y base de la lengua. Piel: hiperpigmentación cutánea (piel bronceada). Faneras: pérdida del vello corporal, facial, pubiano y axilar. (Fig. 2)
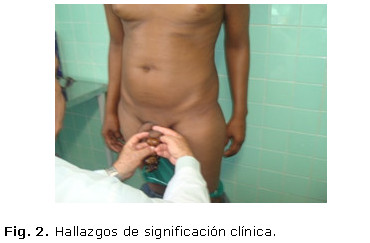
Abdomen: plano suave, depresible, no doloroso a la palpación superficial ni profunda con hepatomegalia palpable de más menos 3 cm por debajo del reborde costal derecho, de ambos lóbulos, no dolorosa, de superficie lisa y bordes romos.
Se indicaron complementarios resultando positivos los siguientes:
Hb: 109g/l; Hierro sérico: 51 mmol/l; AlaninoAminotransferasa (ALAT):78 U/L; Aspartato Aminotransferasa (ASAT) :134 U/L; Gammaglutamiltransfersa (GGT): 65 U/L; Fosfatasa Alcalina (FA): 308 U/L, Proteínas Totales : 54 g/l, Albúmina: 30 g/l, Bilirrubina total en 33 mmol/l con la fracciones directa e indirecta en 19 y 14 mmol/l respectivamente, colesterol: 2,8 mmol/l y glicemia en 7,1 mmol/l. Se realizaron, además determinaciones específicas del hierro, en este caso, el indice de saturación de la transferrina (IST) en 80 % y la ferritina sérica (FS) en 1050ng/ml, resultando ambas elevadas.
Se efectuaron otros exámenes como: coagulograma completo, conteo de plaquetas, leucograma con diferencial, eritrosedimentación, perfil renal, dosificación de ceruloplasmina,alfafeto proteína y amilasa pancreática, todos dentro de valores normales. Resultaron negativos también los marcadores de hepatitis B y C (Antígeno de superficie y Anticuerpo contra hepatitis C).
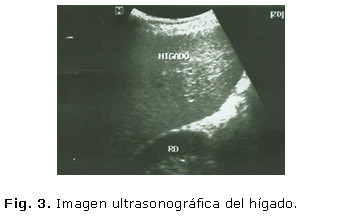
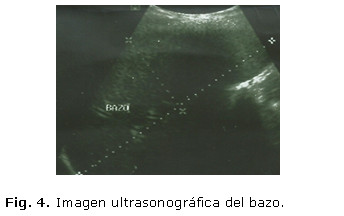
Se realizó ultrasonido abdominal donde se corroboró la hepatomegalia detectada al examen físico, con un discreto aumento de la ecogenicidad hepática y pancreática, sin nódulos visibles ni otras masas tumorales, no adenopatías intrabdominales, ligera esplenomegalia, sin líquido libre en cavidad abdominal ni otras alteraciones ultrasonográficas en el resto de los órganos abdominales: Imagen ultrasonográfica del hígado (figura 3) y el bazo (figura 4).
Se indicó endoscopia del tracto digestivo superior no detectándose várices esofágicas ni otros signos de hipertensión portal, solo una gastritris eritematosa antral ligera.
Con estos resultados y ante la sospecha de una hepatopatía crónica (Inversión del índice ASAT/ALAT(>1), hipoalbuminemia, hiperbilirrubinemia,hipocolesteronemia y hepatomegalia) asociado a anemialigera y evidencia bioquímica de alteraciones en le metabolismo del hierro (hierro sérico, IST y FS elevados), se indicó laparoscopia con biopsia hepática con la impresión diagnóstica de una posible cirrosis hepática por hemocromatosis.
En el examen macroscópico realizado a través de la Laparoscopia, se observó el hígado aumentado de tamaño más menos 4 cm por debajo del reborde costal derecho, de color carmelita oscuro, superficie irregular, de aspecto granular fino con tendencia a la formación de nódulos finos, además múltiples y finos tractus de fibrosis de color blanquecino en las caras anterior e inferior de ambos lóbulos, el borde estaba romo y la consistencia hepática aumentada. Se detectó discreto aumento de la vascularización de los ligamentos falcifome y redondo así como esplenomegalia grado I, sin presencia de liquido libre en cavidad ni alteraciones en el peritoneo parietal ni visceral.
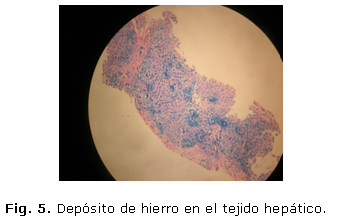
Según estas observaciones se concluyó el caso como hepatopatía crónica en evolución a la cirrosis hepática por posible hemocromatosis, y con este diagnóstico se decidió realizar biopsia hepática para su posterior confirmación. La muestra fue enviada al departamento de Anatomía Patológica del centro y procesada por especialistas del mismo, quienes utilizaron la tinción especial de azul de prusia. El resultado demostró la presencia de depósito de hierro intrahepatocitario y de disposición pericanalicular, en las células de Kupffer y el tejido conjuntivo, además distorsión de la arquitectura hepática. El caso se concluyó como una cirrosis hepática inicial por depósito de hierro, corroborando así el diagnóstico clínico y laparoscópico. (Fig. 5)
Con este resultado se decidió, en ese momento, la atención del paciente por un equipo multidisciplinario, integrado por médicos de los Servicios de Endocrinología, Hematología y Gastroenterología; dada las características del paciente, su edad, la ausencia de otras patologías que justificasen el depósito secundario de hierro en los tejidos.
Se llegó a la conclusión que debía tratarse de un paciente con hemocromatosis hereditaria y con este diagnóstico se remitió a la consulta provincial de Genética para efectuar los correspondientes estudios genético-moleculares, los cuales fueron procesados en el Centro Nacional de Genética por el método de reacción en cadena de polimerasa, seguido por digestión enzimática. Se concluyó el caso como homocigótico para la mutación C282Y (HH tipo I), aún pendiente el estudio familiar.
El paciente comenzó tratamiento con un medicamento quelante del hierro, en este caso la desferoxamina mesilato (Desferal ), BB de 500 mg, el cual se indicó a dosis de 30 mg/Kg, por vía endovenosa, con una frecuencia de dos veces a la semana, en régimen ambulatorio pero bajo la vigilancia estricta de los hematólogos durante cada administración. El mismo se aplicó por un período de dos meses, lográndose así reducir los niveles de hierro en sangre a 25 mmol/l ( un 50 % con respecto a sus cifras iniciales).
En ese momento el paciente comenzó a presentar disnea a los esfuerzos, palpitaciones, tos seca, dolor en hipocondrio derecho, edemas en miembros inferiores, asociado a taquicardia supraventricular paroxística, por lo que se decidió suspensión del tratamiento, luego de ser discutido el caso con el servicio de Cardiología. Fue hospitalizado por esta sintomatología en el servicio de Medicina Interna con la impresión diagnóstica de debut de insuficiencia cardiaca, llevando seguimiento conjunto con Cardiología.
Luego de efectuar los estudios correspondientes para esta complicación se determinó que se trataba de una miocardiopatía, la cual se consideró una complicación de su enfermedad de base, las descompensaciones frecuentes de dicha patología lo han obligado a reingresar en cuatro ocasiones más.
En estos momentos, luego de dos años de diagnóstico de la enfermedad, el paciente se mantiene compensado tanto desde el punto de vista hepático como cardiovascular, llevando adecuadamente su tratamiento higiénico dietético, así como el farmacológico correspondiente para ambas complicaciones. Continúa con vitaminas antioxidantes y sesiones de flebotomías, esta última con una frecuencia semanal o quincenal dependiendo de sus cifras de hemoglobina. Actualmente mantiene valores de hierro en sangre periférica que oscilan entre los 25 y 30 mmol/l. En la endoscopia superior diagnóstica evolutiva realizada hace dos meses aparecieron várices esofágicas grado I, no han aparecido masas tumorales hepáticas según resultados de los ultrasonidos abdominales evolutivos, de igual forma se mantienen dentro de valores normales las cifras de alfafeto proteína en sangre periférica.
DISCUSIÓN
Según lo reportado en la literatura, la hemocromatosis hereditaria tipo Ies la más frecuente de todas las variedades clínico genéticas establecidas (95 %); de las mutaciones estudiadas en el gen HFE, la C282Y ha sido la más identificada,(1-3,7-11) coincidiendo con la diagnosticada en este caso.
Además de las formas clínicas primarias o hereditarias se describe la hemocromatosis secundaria o adquirida,en las que otras patologías, principalmente de origen hematológico, hepático o genético, favorecen la sobrecarga de hierro a nivel tisular.(1,5,10,12,13) Todas ellas, quedaron descartadas en este paciente.
La mayoría de los autores coinciden que en el caso de la HH, aunque el defecto genético existe desde el nacimiento, los signos y síntomas aparecen en la edad adulta, generalmente después de los 40 años, cuando la demasía de hierro es más agresiva.(1,10)
En este caso la edad de presentación fue mucho más temprana. El acumulo progresivo e irreversible del hierro a nivel tisular es el responsable de la larga lista de síntomas y signos que los pacientes pueden experimentar a lo largo de su vida. Según lo revisado del tema en la literatura (1-4,12) y tal como sucedió en este paciente, la astenia y las fatigas son parte de los síntomas iniciales más descritos por los enfermos.
La afectación en el hipotálamo e hipósifis es observada entre el 15 y el 35 % de los casos, la hipofunción gonadal resultante de esta afectación, produce un déficit en la secreción de gonadotropina (hipogonadismo hipogonadotrópico) lo que trae consigo atrofia testicular, pérdida de la libido e impotencia sexual,(1,3,10,13) síntomas que motivaron a este paciente a solicitar ayuda médica especializada.
El hígado representa uno de los órganos dianas en esta patología. El grado de afectación depende de la cuantía del acúmulo del metal en dicho tejido,habitualmente, las manifestaciones clínicas son compatibles con una hepatopatía crónica que en la mayoría de los casos, tal y como pudo apreciarse en este paciente, evolucionan hacia la cirrosis hepática.(1-3,12,14)
La piel constituye otro de los órganos afectados en esta patología (70 % de los casos), la hiperpigmentación cutánea, causado por el acúmulo de hierro, la pérdida del vello pubiano, facial y axilar completan la lista de signos dermatológicos,(1-3,8,10) todos presentes en este paciente.
Los enfermos pueden presentar síntomas y signos indicativos de afectación cardiovascular, los cuales pueden aparecen entre el 15-35 % de los casos según lo reportado en la literatura.(1,10,12-14) La insuficiencia cardiaca es indicadora de enfermedad avanzada y ha de considerarse secundaria a la miocardiopatía, la cual a su vez resulta del depósito del metal en el tejido miocárdico.(1,10,15) Las arritmias pueden ser también parte del cuadro clínico (35-45 %).(1,13) En este paciente se diagnóstico una Insuficiencia Cardiaca Congestiva en el curso precisamente de una Miocardiopatía dilatada con arritmia asociada, luego de dos meses de diagnosticada la hemocromatosis.
Los estudios hematológicos y bioquímicos son cruciales para el diagnóstico Entre las alteraciones más reportadas están el incremento del hierro sérico (HS) y las transaminasas, ambos complementarios positivos este caso. La existencia de un indice de saturación de la transferrina (IST) mayor al 45 % y ferritina sérica (FS) elevada (mayor que 200 ng/mL en el sexo femenino y 300 en el masculino), son fieles indicadores del exceso del mineral en el organismo, por lo que forman parte indiscutible del estudio de estos casos, en este paciente, los valores detectados coincidieron con las cifras esperadas en esta enfermedad.(1-3,10,14)
La elevación de las aminotranferasas, han sido descritas en más del 65 % de los casos y con frecuencia, como ocurrió en este paciente, se observa inversión del índice ASAT/ALAT, indicador de daño hepático crónico con o sin Cirrosis establecida.(1,2,13,14)
Los estudios genéticos moleculares son primordiales para el diagnóstico específico del subtipo de HH, de ahí su valor e indiscutible indicación. (1,3,15)
El estudio histológico del hígado es esencial para determinar la existencia y concentración del hierro en este órgano, evalúa el grado de lesión histológica y resulta muy útil en el diagnóstico diferencial con otras enfermedades hepáticas de comportamiento clínico y bioquímico similar. El método para la visualización directa del metal fue el utilizado en este caso (Azul de Prusia); es ideal determinar el índice hepático de hierro, (no disponible en nuestro medio), valores por encima de 1.9 son altamente sugestivos de hemocromatosis.(5,10)
Según lo señalado en la literatura actual es imprescindible, para establecer el diagnóstico de la HH demostrar: la presencia de sobrecarga de hierro (elevación de IST, FS, y HS), al menos en dos ocasiones separadas por un período mínimo de tres meses, la existencia de alguna de las mutaciones establecidas y el acúmulo del metal sea por biopsia hepática o métodos imagenológicos, pilares todos que se tomaron en consideración para establecer el diagnóstico de certeza en este caso.(1,5,10,13)
En este paciente la enfermedad se diagnosticó antes de los 30 años pero desafortunadamente en estadios avanzados con repercusión importante en varios órganos. De las diferentes complicaciones descritas en la literatura,(1-3,13-15) al mismo le han sido diagnosticadas hasta el momento: la Cirrosis hepática, la Insuficiencia Cardiaca Congestiva secundaria a una miocardiopatía dilatada así como el hipogonadismo hipogonadotrópico, de ellas las dos primeras junto al hepatocarcinoma se consideran las primeras causas de mortalidad para esta enfermedad,(1,2,5,10) lo que apunta hacia un pronóstico realmente muy reservado en este caso.
El objetivo primordial del tratamiento es lograr la depleción de hierro en exceso depositado en los tejidos, para ello se utilizan básicamente dos opciones terapéuticas: las flebotomías o los medicamentos quelantes del hierro,(1,3,11,14) ambas utilizadas en este paciente en diferentes momentos como se señaló en la presentación.(1-3,5,14)
La mayoría de los especialistas son del criterio que las flebotomías realizadas en estadíos iniciales pueden evitar el desarrollo de manifestaciones clínicas, mientras que los casos avanzados se benefician significativamente mejorando su supervivencia y calidad de vida.(3,10,12) Como norma general, todos los individuos con diagnóstico confirmado tienen criterio de recibir esta terapéutica,con ella el hierro sérico disminuye, se estimula la eritropoyesis, logrando de esta forma la movilización tisular del mineral.(10,13,15) Coinciden la mayoría de los autores quela efectividad demostrada, la experiencia acumulada en su uso, la disponibilidad, seguridad y el bajo costo, unido a la posibilidad de mejorar la cirrosis, hacen de esta técnica el pilar principal del tratamiento en la HH.(1,3,5,14)
Los quelantes de hierro serían, siempre que fuese tolerada por los enfermos, la alternativa terapéutica ideal para la HH y han mostrado ser igualmente eficaces en el tratamiento de la Hemocromatosis secundaria.(3,9,14) Aún no se cuenta en Cuba con presentaciones por vía subcutánea u oral (Retard) de mucho más fácil administración que la Deferoxamina (EV) disponible en nuestro medio, que fue la empleada en este paciente.(3,5,8,10)
La hemocromatosis debe ser, sin lugar a dudas, una de las enfermedades a considerar en el estudio de todo paciente con o sin hipogonadismo hipogonadotrópico post-puberal, en el que se detecten signos clínicos y alteraciones hematológicas y bioquímicas indicativos de hepatopatía crónica. Es necesaria la confirmación diagnóstica de la enfermedad mediante complementarios que evidencien trastornos en el metabolismo del hierro, el examen histológico que demuestre el depósito tisular de este metal y el correspondiente estudio genético para determinar el subtipo de HH. La inmediatez en el diagnóstico y la selección del tratamiento adecuado, impedirán en estos pacientes la presentación de las disímiles complicaciones que forman parte indiscutible de la evolución clínica de esta enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1- Castañeda Guillot C, Clark Feoktistova Y, Ruenes Domech C, Robaina Jiménez Z. Trastornos metabólicos y hereditarios del hígado. En: Paniagua Estévez ME, Piñol Jiménez FN. Gastroenterología y Hepatología clínica. Tomo 7 [Internet]. La Habana: Ecimed; 2015. [citado 11 Ene 2016]. p. 358-71. Disponible en: http://www.bvs.sld.cu/libros/gastroenterologia_hepatologia_tomo6/cap_158.pdf
2- Fernández Delgado ND, Forrellat Barrios M, Valledor Tristá R, et al. Hemocromatosis hereditaria tipo I: a propósito de cuatro casos confirmados. Rev Cubana Hematol Inmunol Hemoter [Internet]. 2014 [citado 11 Ene 2016];30(1):59-67. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892014000100008
3- Cervera García IA, García Heredia M, Collazo Mesa T. Introducción del diagnóstico molecular de la hemocromatosis tipo 1 en Cuba. Rev Finlay [Internet]. 2013 [citado 11 Ene 2016];3(2):[aprox. 6 p.]. Disponible en: http://revfinlay.sld.cu/index.php/finlay/article/view/119
4- Castiella A, Zapata E, Zubiaurre L, et al. Impact of H63D mutations, magnetic resonance and metabolic syndrome among outpatient referrals for elevated serum ferritin in the Basque Country. Ann Hepatol. 2015 May-Jun;14(3):333-9. Citado en PUbMed; PubMed PMID: 25864213.
5- Lima Santos PCJ, Krieger JE, Pereira AC. Molecular Diagnostic and Pathogenesis of Hereditary Hemochromatosis. Int J Mol Sci [Internet]. 2012 [citado 11 Ene 2016];13(2):1497-511. Disponible en: http://www.mdpi.com/1422-0067/13/2/1497
6- Lima Santos PCJ, Dinardo CL, DelfiniCançado R, et al. Non-HFE hemochromatosis. Rev bras hematol hemoter [Internet]. 2012 [citado 11 Ene 2016];34(4):311-16. Disponible en:http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1516-84842012000400019&lng=en
7- Cançado RD, Chiattone CS. Current approach to hereditary hemochromatosis. Rev bras hematol hemoter [Internet]. 2010 [citado 11 Ene 2016];32(6):469-75. Disponible en: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1516-84842010000600011&lng=en&nrm=iso&tlng=en
8- Adams PC, Barton JC, Guo H, et al. Serum ferritin is a biomarker for liver mortality in the Hemochromatosis and Iron Overload Screening Study. Ann Hepatol. 2015 May-Jun;14(3):348-53. PubMed PMID: 25864215.
9- Martins JM. Universal iron fortification of foods: the view of a hematologist. Rev bras hematol hemoter [Internet]. 2012 [citado 11 Ene 2016];34(6):459-63. Disponible en: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1516-84842012000600017&lng=en&nrm=iso&tlng=en
10- Bacon BR, Adams PC, Kowdley KV, et al. Diagnosis and management of hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology [Internet]. 2011 [citado 11 Ene 2016];54(1):328-43. Disponible en: http://dx.doi.org/10.1002/hep.24330
11- Fleming RE, Ponka P. Iron Overload in Human Disease. N Engl J Med [Internet]. 2012 [citado 4 Feb 2016];366(4):348-59. Disponible en: http://www.nejm.org/doi/full/10.1056/NEJMra1004967
12- Muñoz M, García-Erce JA, Remacha AF. Disorders of iron metabolismo. Part II: iron deficiency and iron overload. J Clin Pathol [Internet]. 2011 [citado 4 Feb 2016];64:287-96. Disponible en: http://jcp.bmj.com/content/64/4/287.full.pdf+html
13- Arancibia JP. Sobrecarga de hierro y enfermedad hepática: aspectos clínicos y terapéuticos. Gastroenterol latinoam [Internet]. 2011 [citado 4 Feb 2016];22(2):152-5. Disponible en: http://sociedadgastro.cl/?option=com_k2&view=item&task=download&id=1285&Itemid=144
14- Cambruzzi E, Pêgas KL, Zettler CG, et al. Hemocromatose associada a carcinoma hepatocelular e neoplasias extra-hepáticas. Rev AMRIGS [Internet]. 2012 [citado 4 Feb 2016];56(1):67-70. Disponible en: http://www.amrigs.com.br/revista/56-1/0000095683-13_796.pdf
15- Bardou-Jacquet E, Lainé F, Deugnier Y. Reply to: "Reduced mortality due to phlebotomy in moderately iron-loaded HFE Haemochromatosis? The need for clinical trials". J Hepatol. 2015 Jul;63(1):283-4. doi: 10.1016/j.jhep.2015.03.030. Epub 2015 Apr 1. PubMed PMID: 25841362.
Recibido: 8 de abril de 2016.
Aceptado: 7 de junio de 2016.
Sahilí Corrales Alonso. Hospital Universitario Clínico Quirúrgico Comandante Faustino Pérez Hernández. Carretera Central. Km. 101 Matanzas, Cuba. Correo electrónico: sahili.mtz@infomed.sld.cu

