










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Informática Médica
versión On-line ISSN 1684-1859
RCIM vol.8 supl.1 Ciudad de la Habana 2016
ARTÍCULO ORIGINAL
Similitud molecular empleando Índices Híbridos
Molecular similarity using Hybrid Indices
Aurelio Antelo-Collado,I Juan L. Paneque-Pérez,II María C. Hernández-Govea,III Ramón Carrasco-VelarIV
I Universidad de las Ciencias Informáticas, Carretera a San Antonio de los Baños km 2½, La Habana, Cuba. E-mail: aantelo@uci.cu
II Universidad de las Ciencias Informáticas, La Habana, Cuba. E-mail: jlpaneque@uci.cu
III Universidad de las Ciencias Informáticas, La Habana, Cuba. E-mail: mcgovea@uci.cu
IV Universidad de las Ciencias Informáticas, La Habana, Cuba. E-mail: rcarrasco@uci.cu
RESUMEN
Se presenta un método para la detección de semejanza entre moléculas basado en macheo inexacto de grafos. Se parte del grafo molecular completo ponderado en sus vértices por propiedades químico-físicas particionadas sobre los mismos, se reduce el grafo por el procedimiento CALEDE que define Centros Descriptores o fragmentos de primer orden, los cuales son subgrafos ponderados por la suma de los valores de los vértices ponderados individualmente a su vez, y se construyen fragmentos denominados de segundo orden que incluyen la distancia entre los centros de masas de ambos centros descriptores. Se presenta el método de búsqueda aplicado a una base de datos de más de 300 moléculas con sus respectivas estructuras en tres dimensiones. Esos compuestos se encuentran evaluados como anticancerígenos en la base de datos de compuestos del NCBI-USA. En el experimento computacional se encuentra que, en dependencia de la función de similitud empleada, es posible detectar compuestos que a pesar de poseer diferente topología, poseen valores de las propiedades empleadas para el macheo lo cual sugiere la presencia de potenciales farmacóforos como hallazgo relevante, lo cual constituiría un novedoso enfoque para el diseño computacional de fármacos.
Palabras Clave: similitud molecular, índices híbridos, CALEDE.
ABSTRACT
A method for detecting similarity between molecules based on inexact matching graph is presented. We start from a complete molecular graph vertices weighted by several hybrid indices. The molecular graph is reduced by CALEDE procedure, which define descriptors centers or first order fragments. These fragments are subgraphs weighted with the sum of values of the vertices weighted with the hybrid indices. It also define second order fragments by including the distance between the centers of mass of both descriptors centers. The search method applied to a database of over 300 molecules with their respective threedimensional structures is presented. These compounds are reported in the NCBI-USA database of compounds whish were evaluated in anticancer tests. In the computational experiment, depending on the similarity function used, is possible to detect compounds that despite having different topology have property values suggesting the presence of potential pharmacophore. It suggest the possibility to use this approach as a novel approach for computational drug design.
KeyWords: molecular similarity, hybrid índices, CALEDE.
INTRODUCCIÓN
Se ha demostrado que los índices híbridos1 propuestos por Carrasco y cols. para la descripción simultánea de propiedades químico-físicas y la estructura química permiten describir y representar una molécula o fragmento de ella de tres formas diferentes utilizando un procedimiento computacional y matemáticamente sencillo. Permite también comparar estructuras diferentes a partir de la utilización de dichos índices, lo cual ha abierto ha abierto una nueva puerta para el trabajo de búsquedas intensas en bases de datos.
En las bases de datos de moléculas orgánicas es posible encontrar grandes diferencias estructurales. Cuando el objeto de la base es el almacenamiento de moléculas biológicamente activas, se encuentra entonces que la elevada diferencia estructural entre los compuestos revela dos diferentes paradojas estructurales; compuestos estructuralmente semejantes presentan actividades distintas, o compuestos estructuralmente diferentes presentan similar actividad. Con los nuevos descriptores híbridos se pretende contribuir a la resolución de este problema.
Son múltiples y diferentes los métodos para la realización de búsquedas en bases de datos de estructuras químicas. La utilización de medidas de semejanza o similitud entre grafos (macheo exacto de grafos), es uno de los procedimientos más empleados históricamente en esta área de la química grafo teórica. En el presente trabajo se presentan los procedimientos desarrollados para el establecimiento de los criterios de similitud diferencia entre fragmentos o moléculas utilizando los índices híbridos, en un macheo inexacto de grafos.
SOFTWARE Y PROCEDIMIENTOS
Datos
El conjunto de datos de trabajo es el ensayo AID941 del NCBI.2 Está formado por un conjunto de 330 moléculas. El formato de fichero de datos estructurales utilizado de las moléculas fue el *.mol o *.sdf. Las estructuras químicas se visualizan empleando el Jmol, visualizador de Java de código abierto que realiza la representación gráfica tridimensional de alto rendimiento sin requerimientos de hardware, ya que solo precisa de la instalación de la Máquina Virtual de Java.
Descriptores topográficos
Como descriptores topográficos a utilizar se emplearon los índices de Estado Refracto, Electro y Lipotopográfi-cos para Átomos desarrollados por Carrasco y cols.1 Para el cálculo de estos índices se utilizó la librería Chemistry Development Kit (CDK),3 una librería de código abierto programada en Java que está destinada para la realización de cálculos en química computacional, la quimio y la bioinformática. La reducción del grafo molecular se realizó con el algoritmo CALEDE,4 definido por Carrasco y cols. e implementado en la librería CDK. Mediante CALEDE se generan fragmentos moleculares de segundo orden definidos como dos Centros Descriptores (CD) más la distancia euclidiana entre los centros de masas de los respectivos CD. Los Centros Descriptores de CALEDE (fragmentos de primer orden) son agrupaciones típicas de grafos como ciclos o anillos, estrellas de orden 3 y 4, los halógenos, oxígeno, nitrógeno y los grupos metilo, metileno y metino.
Funciones de similitud
Como funciones de similitud se emplearon las de Sørensen, Dice-Sørensen, Tanimoto, Jaccard, Ruzicka, Cze-kanowski y Soergel, incluidas en la revisión de Sung5 por considerarse que son las que mejor se adaptan al cálculo de similitud molecular, basados en vectores de valores.
RESULTADOS Y DISCUSIÓN
Cálculo de los umbrales de similitud a utilizar
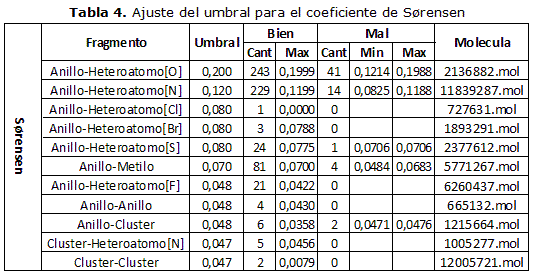
Un elemento a tener presente cuando se realizan cálculos de semejanza mediante el empleo de funciones de similitud, lo constituye el grado de error permisible que se utilizará. Es por ello, que antes de realizar las pruebas para validar los algoritmos implementados, se hace indispensable determinar el umbral de máxima diferencia o valor de mínima similitud de cada una de las funciones seleccionadas, el cual se estará empleando en las búsquedas que se realizarán y constituirán las propuestas a umbrales de esta investigación, aunque el criterio de determinación puede cambiar, por lo que queda a decisión del investigador establecer el umbral que desee para sus experimentos. Para determinar los umbrales se escogieron arbitrariamente once fragmentos moleculares de segundo orden, pertenecientes a moléculas presentes en el ensayo AID9416, con los cuales se realizaron búsquedas para ajustar los límites deseados, cada uno de los fragmentos seleccionados se examinaron con los fragmentos presentes en cada una de las 330 moléculas contenidas en el propio ensayo AID941. En los experimentos realizados se considera un resultado correcto cuando los fragmentos obtenidos pertenecen al mismo tipo de fragmento seleccionado e incorrecto en caso contrario.
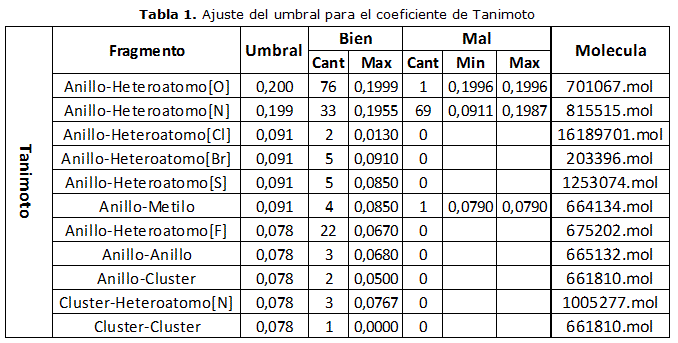
En la tabla 1 se muestra el procedimiento realizado para el coeficiente de Tanimoto. Inicialmente se realiza la búsqueda para el fragmento compuesto por un anillo y un heteroátomo de oxígeno, tomando un umbral lo suficientemente amplio, para que se obtuvieran resultados correctos e incorrectos, obteniéndose 76 fragmentos del mismo tipo del fragmento seleccionado y 1 de otro tipo (con un valor de 0.1996 de diferencia), por lo que es necesario disminuir el umbral hasta 0.199. Con este nuevo umbral, se realiza una nueva búsqueda, tomando un fragmento compuesto por un anillo y un heteroátomo de nitrógeno, obteniéndose 33 bien y 69 mal (con valores entre 0.0911 y 0.1987 de diferencia), por lo que se hace necesario un nuevo ajuste del umbral de forma tal que sea menor que el menor valor de diferencia obtenido dentro de los resultados incorrectos, fijándose el nuevo umbral hasta 0.091. A continuación, y con este nuevo valor, se realiza nuevamente la búsqueda, pero con un fragmento compuesto por un anillo y un heteroátomo de cloro, encontrándose 2 fragmentos correctos, por lo que no es necesario disminuir el umbral. Este procedimiento se realiza para cada uno de los 7 restantes fragmentos, modificando el valor si se encuentran resultados incorrectos y manteniéndolo igual en otro caso.
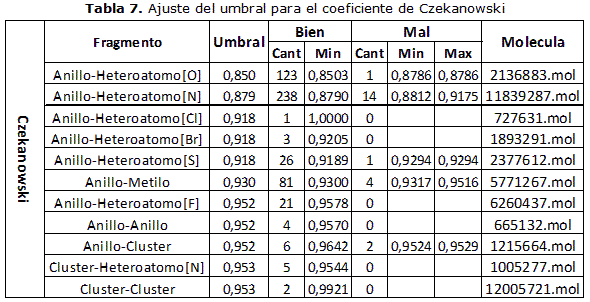
El método descrito anteriormente se realizó para cada una de las funciones de similitud y de distancia seleccionadas para esta investigación, tomando como base los fragmentos seleccionados. Cabe destacar que en el caso de las funciones de similitud el ajuste de umbral se realizó tomando un valor mayor que el mayor valor de similitud obtenido dentro de los resultados incorrectos y en el caso de las funciones de distancia, como se explicó anteriormente en el caso del coeficiente de Tanimoto. En las tablas 2-3- 4-5-6-7 se muestran los valores obtenidos en los experimentos realizados.




En la tabla 8 se presenta el resumen de los umbrales para cada una de las 7 funciones de similitud/distancia seleccionadas para esta investigación.
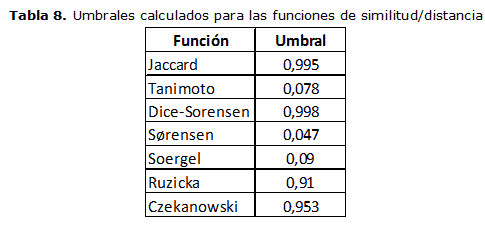
Es necesario aclarar que los valores de umbral calculados, fueron obtenidos a partir del algoritmo de búsqueda de fragmentos simples implementado y teniendo en cuenta las tres propiedades químico-físicas para cada uno de los fragmentos analizados, estos valores pueden cambiar para las búsquedas en las que solo se tenga en cuenta uno de las propiedades.
Utilización de la fragmentación y los índices híbridos
No todas las moléculas cumplen el principio de Johnson y Maggiora4 de que compuestos estructuralmente semejantes exhiben propiedades semejantes. Como contradicción a este postulado existen las paradojas estructurales, expresadas en que moléculas similares estructuralmente tienden a exhibir propiedades biológicas diferentes y moléculas diferentes estructuralmente tienden a exhibir propiedades biológicas similares. Por lo antes planteado, el cotejo exacto de grafos no constituye una elección a la hora de realizar búsquedas de similitud molecular. La necesidad de realizar comparaciones entre moléculas y encontrar un método para determinar similitud molecular, nos condujo al empleo de descriptores híbridos ponderados por propiedades químico-físicas, específicamente el índice de estado Refractotopográfico para átomos, el índice de estado Electrotopográfico para átomos y el índice de estado Lipotopográfico para átomos, los cuales reportados anteriormente.
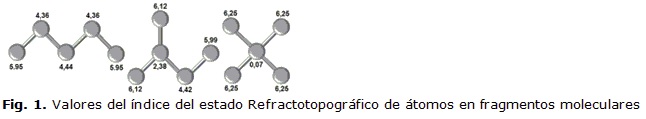
En la figura 1 se muestra el comportamiento del valor del índice del estado refractotopológico. Puede verse la diferencia de valor del índice, dada la diferente distribución espacial de estos compuestos, los cuales son considerados isómeros de posición por su diferente distribución en la estructura. Obsérvese como los átomos externos varían su valor de una estructura a la otra y como disminuye apreciablemente el valor del átomo situado geométricamente al centro. Esta variación está dada por la influencia que ejercen el resto de los átomos sobre los restantes de la molécula.
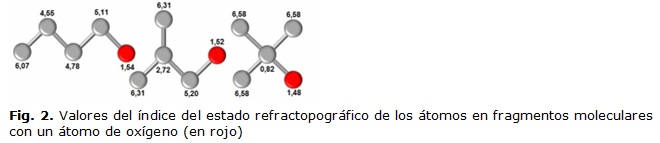
Otro ejemplo que ilustra lo planteado anteriormente lo constituyen los fragmentos mostrados en la figura 2, donde se muestra que los valores del Índice del Estado Refractotopográfico de los átomos que conforman a los fragmentos moleculares varían al cambiar uno de los átomos presentes en los mismos, evidenciándose que el índice revela el comportamiento de las propiedades químico-físicas y cómo esta varía, no solo en dependencia del tipo de átomo y de la estructura que presenten los fragmentos, sino del efecto que ejercen los átomos vecinos. Este hecho sugiere el empleo de los índices para estudios de similitud molecular.
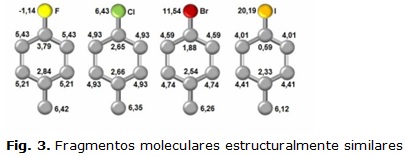
En la figura 3 se muestran fragmentos estructuralmente iguales, que difieren debido a la existencia de diferen-tes átomos pesados (F, Cl, Br, I), cuyos valores del índice del estado refractopográfico son distintos, y la in-fluencia de cada uno de estos sobre el resto provoca que estos presenten valores diferentes en sus propieda-des, consecuentemente presentan actividad biológica diferente. Por lo antes planteado el empleo de los índices de Carrasco y cols. constituye uno de los ejes de esta investigación.
Otro elemento fundamental lo constituye la fragmentación molecular utilizando los centros descriptores de CA-LEDE. Como se aprecia en la tabla 9, los subgrafos mostrados son topológicamente idénticos y se han definido como fragmentos de segundo orden al estar formados por un anillo y un heteroátomo, excepto el cuarto que está formado por un anillo y un metilo.
Debe acotarse que las propiedades químicas de un grupo metilo difieren apreciablemente a las de un heteroátomo, aunque sea comparable en cuanto a tamaño con respecto al bromo o el iodo.
Otro de los criterios que permite validar la utilización de la fragmentación propuesta, en los estudios de similitud molecular se evidencia en la tabla 10.
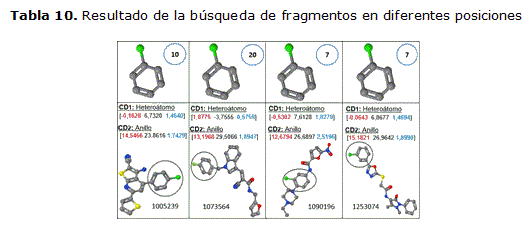
En la misma se muestra la realización de cuatro búsquedas tomando varios fragmentos que estructuralmente son idénticos, pero como se evidencia las propiedades químico-físicas de cada uno de ellos son diferentes, este cambio se debe fundamentalmente a la posición espacial con respecto al resto de los átomos que conforman a las moléculas a las cuales pertenecen, debido a que en algunas el fragmento se encuentra en un extremo y en otros casos se encuentra al centro, por lo que la influencia sobre el fragmento especifico varia en los diferentes casos.
En la tabla se muestra la cantidad de resultados encontrados para cada una de las posiciones, según se muestra en la molécula en la parte inferior de la tabla.
Los criterios planteados anteriormente sugieren que para realizar estudios de similitud molecular no se debe tener en cuenta solamente métodos que trabajen a partir de la estructura de las moléculas, sino que es razonablemente aconsejable, explorar alternativas como la propuesta en este trabajo, mediante el empleo de una forma de fragmentación de las moléculas, que aporte información no solo estructural, sino además de propiedades asociadas a esas estructuras como la que brindan el algoritmo CALEDE de fragmentación y los descriptores híbridos
ponderados con propiedades químico físicas particionadas sobre los vértices del grafo molecular.
Búsquedas de fragmentos simples
Los fragmentos de segundo orden son, como se explicó anteriormente, los subgrafos del grafo reducido de CALEDE que están formados por dos Centros Descriptores. A partir de estos fragmentos se forma un vector formado por los valores de los índices Electrotopográfico, Refractotopográfico y Lipotopográfico de cada Centro Descriptor y la distancia entre los centros de masas de cada uno de ellos. Esta representación en vectores de números reales, permite la aplicación de funciones matemáticas, que determinen la semejanza entre ellos.
En la tabla 11 se muestra el fragmento A6-HCl1 perteneciente a la molécula 12005721 del ensayo AID941, en la misma aparecen los valores de los y la distancia entre los centros descriptores que lo forman.
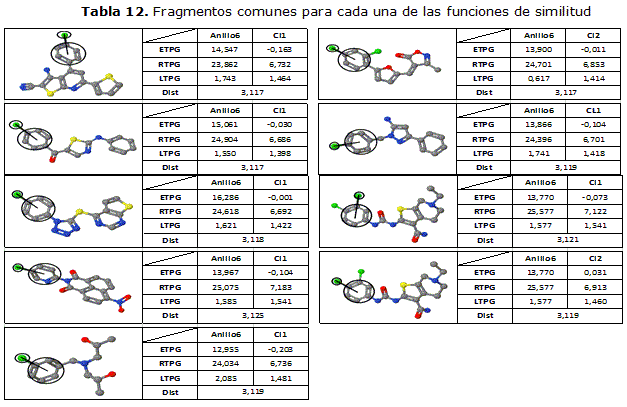
Tomando como objetivo el fragmento mostrado en la tabla 11, se realizaron varias ejecuciones del algoritmo implementado para la búsqueda de fragmentos similares aplicando cada una de las funciones de similitud seleccionadas. En la tabla 12 se muestran los fragmentos en común que fueron encontrados por cada una de las funciones de similitud.
A partir del análisis de los resultados, se evidencia que el algoritmo implementado permite encontrar aquellos fragmentos dentro del ensayo que poseen gran semejanza en los valores de los índices con el fragmento objetivo y a la vez presentan una notable semejanza estructural.
En la figura 4 se muestra la cantidad de resultados devueltos por el algoritmo, por cada una de las funciones de similitud seleccionadas.
De las nueve funciones de similitud evaluadas, solo Jaccard y DiceSorensen devuelven un menor número de fragmentos. El procedimiento aquí presentado introduce el enfoque del macheo inexacto de grafos moleculares para grafos ponderados por propiedades quimico-físicas particionadas, lo cual puede resultar una nueva vía de aproximación a la detección de regiones de moléculas involucradas directamente en la respuesta biológica, determinado por algunas propiedades químico-físicas, con lo cual se abriría una nueva opción para el diseño computacional de fármacos.
CONCLUSIONES
Se desarrolló un procedimiento de búsqueda de fragmentos moleculares basado en el método CALEDE de fragmentación de moléculas ponderadas por los índices topográficos híbridos de Carrasco et.al. y el principio del macheo inexacto de grafos. El empleo de diferentes funciones de similitud mostró la factibilidad del método empleado para extraer información de diferente naturaleza de bases de datos de estructuras químicas.
AGRADECIMIENTOS
Los autores, agradecen las facilidades brindadas por la Universidad de Ciencias Informáticas para la realización de este trabajo.
REFERENCIAS BIBLIOGRÁFICAS
1. Carrasco-Velar R, Prieto-Entenza J.O, Antelo-Collado A, Padrón-García J.A, Cerruela-García G, Maceo-Pixa Á.L, Alcolea-Núñez R, Silva-Rojas L.G. (2013). Hybrid reduced graph for SAR studies, SAR and QSAR in Environmental Research. DOI:10.1080/1062936X.2013.764926.
2. NCBI. PubChem BioAssay Database AID941. Disponible en: http://www.ncbi.nlm.nih.gov/pcassay
3. Steinbeck Christoph Han, Yongquan Kuhn S, Horlacher O, Luttmann E, Willighagen E. The Chemistry Development Kit (CDK): an open source Java library for Chemo and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003. 43 (2), pp 493-500.
4. Carrasco-Velar R, Trinchet-Almaguer D, Ortiz-Tornin S, Pérez-Durán R.E. CALEDE Lenguaje descriptor de la estructura química. QUITEL XXXIII.
5. Sung Hyuk, Cha. Comprehensive Survey on Distance/Similarity Measures between Probability Density Functions, 2007.
Recibido: 22 de marzo de 2016.
Aprobado: 12 de mayo de 2016.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}