










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
Medisur vol.10 no.1 Cienfuegos ene.-feb. 2012
PRESENTACIÓN DE CASO
Síndrome de Rett. Presentación de un caso
Rett Syndrome. A Case Presentation
Julio Padrón GonzálezI , Ramón Pérez MejíasII , Lidys Padrón FernándezI , Leydis Padrón FernándezIII
I Univerisdad de Ciencias Médicas, Cienfuegos, Cienfuegos, Cuba, CP: 55100
II Policlínico Universitario. Área 3, Cienfuegos, Cienfuegos, Cuba
III Policlínico Universitario. Área 7, Cienfuegos, Cienfuegos, Cuba
RESUMEN
El síndrome de Rett es un trastorno neurológico de base genética. Afecta casi exclusivamente a niñas y mujeres; la incidencia estimada en la población general es de un caso por cada 10.000 mujeres, en su tipo clásico, es de 1 por cada 15. 000 nacimientos. Su diagnóstico es descriptivo, basado en un conjunto de signos y síntomas, pero no es etiológico; el tratamiento es sintomático y de apoyo. La enfermedad, frecuentemente, suele estar mal diagnosticada como autismo o parálisis cerebral. Debe sospecharse en pacientes del sexo femenino, con diagnóstico de parálisis cerebral infantil o retardo mental idiomático, apoyado en criterios establecidos internacionalmente. Se presenta el caso de una niña de 11 años de edad, visitada en el hogar por médicos de la Misión Barrio Adentro en la República Bolivariana de Venezuela, la cual fue normal hasta aproximadamente los 2 años, cuando comenzó con deterioro en sus destrezas psicomotoras, alteración social y de conducta con autismo infantil, crisis epilépticas frecuentes y retardo mental importante.
Palabras clave: síndrome de Rett, informes de casos, Venezuela.
ABSTRACT
Rett syndrome is a neurological disorder of genetic basis. It affects almost exclusively girls and women being the estimated incidence of this disease in the general population of one case per 10,000 women. In its classical type it is of one in every 15. 000 births. Its diagnosis is descriptive, based on a set of signs and symptoms, but not etiologic. Treatment is symptomatic and supportive. Rett syndrome is frequently misdiagnosed as autism or cerebral palsy. It should be suspected in female patients, diagnosed with child cerebral palsy or idiomatic mental retardation, supported by internationally established criteria. The case of an 11 years old girl visited at her home and attended by doctors of the Barrio Adentro mission in the Bolivarian Republic of Venezuela is presented. The patient was normal until about two years old when she began presenting impairment of psychomotor skills, social and behavioral disorders with infantile autism traits, seizures and considerable mental retardation.
Key words: Rett syndrome, case reports, Venezuela.
INTRODUCCIÓN Andrés Rett, pediatra de la Universidad de Viena, fue quien reportó por primera vez, en 1966, en Alemania, 31 niñas que habían desarrollado regresión mental en edades tempranas de la vida. (1) En 1978, Ishikawa en Japón, y Hagberg en 1980 en Inglaterra, reportaron casos con síntomas similares a los descritos por Rett. (2) Coronel Carvajal, en una revisión sobre este síndrome, expresa que en noviembre de 1983 se reportó en una revista de neurología una serie de 35 casos de Suecia, Portugal y Francia, y ya se definía como síndrome de Rett. (3) En México se describió el primer grupo de casos en 1989. (4) Este síndrome afecta a todas las razas y es la segunda causa más común de retraso mental grave en el sexo femenino (después del síndrome de Down); ha sido reportado en más de 40 países en todo el mundo. Cuando se incluye el espectro completo del síndrome tiene una incidencia estimada en la población general de un caso por cada 10.000 mujeres; cuando se restringe el tipo clásico, su incidencia es de uno por cada 15.000 nacimientos de niñas vivas. Estudios epidemiológicos suecos han sugerido un prevalencia de 1 por 10.000, pero investigaciones más recientes en Noruega e Italia muestran tazas de 2 por 10.000. (5) En la actualidad, los estudios epidemiológicos de retraso mental indican que existe una prevalencia en la población infantil de 2-3 %. De ellos, en el 30 % de los casos se desconoce la causa y consideran idiopático lo que en realidad pudiera ser la expresión de un síndrome de Rett. (5) El diagnóstico suele ser difícil, ya que comparte características con varias enfermedades psiquiátricas y degenerativas. (5) Se lleva a cabo por la observación y la valoración clínica, al no existir un marcador bioquímico, morfológico, neurofisiológico, citogenético o molecular que facilite la determinación del síndrome. (6) Debe sospecharse en pacientes del sexo femenino, con diagnóstico de parálisis cerebral infantil o retardo mental idiomático, al apoyarse en criterios establecidos internacionalmente en 1988 que incluyen criterios necesarios, complementarios y componentes de exclusión. La identificación de mutaciones en los genes MECP2 y CDKL5 aporta una confirmación genética del diagnóstico clínico. (8) Por lo poco común de la afección y las complicaciones para su diagnóstico se decidió la publicación de este caso. PRESENTACIÓN DEL CASO Paciente de 11 años de edad, femenina, que fue visitada en el hogar por médicos de la Misión Barrio Adentro en la República Bolivariana de Venezuela como parte del proceso de dispensarización de las familias en 2003. La madre estaba fallecida, por lo que se encontraba bajo el cuidado y tutoría de una medio hermana mayor que tiene dos hijos menores y es ama de casa, con bajo nivel cultural y sin apoyo económico; no existen vínculos afectivos con el padre. Refirió la hermana que a partir de aproximadamente los dos años de edad comenzó a dejar de hablar, a no caminar, con cuadros frecuentes de vómitos, respiración agitada y convulsiones, durante las cuales, en ocasiones, pierde el control de esfínteres. Examen físico: Se observó sentada, aseada, con los brazos flexionados frente a la barbilla, con movimientos esteriotipados continuos del cuerpo y de las manos, característicos de retorcerse o lavarse las manos. Llamó la atención la cabeza en extensión, posición que preferentemente ocupa. (Figura 1).
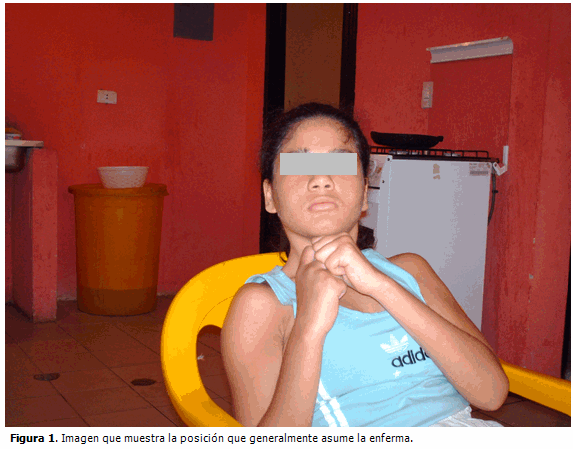
Aparato cardiovascular: Sin alteraciones aparentes. FC: 76/min
Aparato respiratorio: a la inspección se observaron episodios de hiperventilación. Murmullo vesicular conservado, no estertores. FR: 28/min
Aparto digestivo: sialorrea, protusión de la lengua.
Soma: se apreció pie pequeño, hipotrofia muscular en los cuatro miembros, más marcada en los inferiores y escoliosis.
Sistema Nervioso Central: la paciente estaba alerta, sonreía y hacía contacto visual, obedecía órdenes, pero no podía pronunciar claramente las palabras. Ocasionalmente se frotaba las manos como si las estuviese lavando, con movimientos continuos de bamboleo de la cabeza, tronco y de las manos que por momentos cesaban. Fuerza muscular disminuida en los cuatro miembros. Hiperreflexia global, sensibilidad normal. Se sostenía de pie con ayuda; presentaba marcha espástica. El tono muscular estaba aumentado, con signo de rueda dentada, predominantemente en las extremidades izquierdas. Presentaba hipotrofia de las 4 extremidades, fundamentalmente en los miembros inferiores. Presentaba adicionalmente ataxia, dismetría y disinergia de las cuatro extremidades.
Circunferencia cefálica: 49 cm
Electrocardiograma: mostró ritmo sinusal.
Electroencefalograma: mostró lentificación de su actividad de base, reducción de la fase REM del sueño y descargas paroxísticas.
Entrevista a vecinos: hablaron de cómo la niña comenzó a perder sus habilidades manuales y a dejar de hablar lo que fue progresando con el tiempo hasta quedar postrada en la silla. Expresaron que la han llevado a distintos médicos pero nunca le han dado un diagnóstico, que la familia necesita ayuda y que a la niña en particular le hace mucha falta una silla de ruedas.
DISCUSIÓN
En el caso que se presenta en este artículo, la paciente cumplía todas las características clínicas de la forma típica del síndrome de Rett en su estadio 2: desarrollo psicomotor aparentemente normal hasta el segundo año de vida, cuando se inicia una regresión de las funciones cerebrales manifestada por pérdida de las destrezas motoras de las manos, asociada a estereotipias manuales, dispraxia de la marcha y pérdida de las habilidades en la comunicación verbal y no verbal, desaceleración del crecimiento craneal que origina microcefalia, con episodios de hiperventilación, aerofagia y crisis epilépticas. La falta de un diagnóstico y la complejidad del caso hizo que el equipo de médicos internacionalistas se diera a la tarea de revisar toda la literatura posible y realizar los exámenes complementarios a su alcance con el objetivo de llegar a un diagnóstico definitivo, el cual resultó como síndrome de Rett por ser la segunda causa más común de retraso mental grave en el sexo femenino (después del síndrome de Down) y cumplir los criterios necesarios, de apoyo y de exclusión, establecidos internacionalmente. Este planteamiento diagnóstico fue posteriormente confirmado por especialistas cubanos y venezolanos.
Dadas las dificultades que se presentaron para llegar al diagnóstico definitivo de esta paciente, los autores consideran que nuestros médicos generales y pediatras no están familiarizados con sus manifestaciones clínicas, por lo que puede escaparse o equivocarse su diagnóstico. El objetivo de este trabajo es alertar sobre la existencia de la enfermedad y su forma de diagnóstico.
REFERENCIAS BIBLIOGRÁFICAS
1. Pineda M, Aracil A, Vernet A, Espada M, Cobo E, Arteaga R, et al. Estudio del síndrome de Rett en la población española. Rev Neurol. 1999;28(1):105-9
2. Calderón González R, Calderón Sepúlveda RF, Treviño Welsh J. Fenomenología clínica del síndrome de Rett. Gac Med Mex. 1999;135(1):11-8
3. Coronel Carvajal C. Síndrome de Rett: un nuevo reto para los pediatras. Revisión bibliográfica. Rev Cubana Pediatr [revista en Internet]. 2002 [citado 5 Feb 2012];74(2):[aprox. 7p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312002000200010
4. Calderón R, Gramajo O, Sevilla R, Carrera JP, Peña F, Bolaños G. Síndrome de Rett: una causa frecuente, poco reconocida de retraso mental. Rev Mex Ped. 1989;56:191-200
5. Blanco NM, Manresa VS, Mesch GJ, Melgarejo MJ. Síndrome de Rett: criterios diagnósticos. Revista de Posgrado de la VIa Cátedra de Medicina. 2006;(153):22-8
6. Nieto-Barrera M, Nieto-Jiménez M, Díaz Fernández F, Campaña Marchal C, Sánchez ML, et al. Evolución de las crisis epilépticas en el síndrome de Rett. REV NEUROL. 1999;28(5):449-53
7. Campos-Castello J, Fernández-Mayoralas DM, Muñoz-Jareño N, San Antonio-Arce V. Síndrome de Rett: 50 años de historia de un trastorno aún no bien conocido. Medicina(B. Aires) [revista en Internet]. 2007 [citado 5 Feb 2012];67(6-1 suppl 1):[aprox. 6p]. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S0025-76802007000700002
8. Roche-Martínez A, Gerotina E, Armstrong-Morón J, Sans-Capdevila O, Pineda M. FOXG1, un nuevo gen responsable de la forma congénita del síndrome de Rett. REV NEUROL. 2011;52(10):597-602
Recibido: 25 de octubre de 2011.
Aprobado: 21 de febrero de 2012.
Julio Padrón González. Especialista de II Grado en Medicina General Integral. MSc. en Educación Médica. Profesor Asistente. Universidad de Ciencias Médicas. Cienfuegos. Correo electrónico: jpadron@ucm.cfg.sld.cu

