










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
Medisur vol.16 no.5 Cienfuegos set.-oct. 2018
ARTÍCULO ORIGINAL
Diagnóstico molecular de distrofia muscular de Duchenne/Becker en una familia sin antecedentes patológicos de la enfermedad
Molecular Diagnosis of Duchenne/Becker muscular dystrophy in a family with no pathological antecedents of the disease
Ivonne Martín Hernández , Alejandro Ariosa Olea , Mariesky Zayas Guillot , Tatiana Zaldívar Vaillant , Celia Rosa Soto Pérez-Stable
Instituto de Neurología y Neurocirugía, La Habana, Cuba
RESUMEN
Fundamento: Las distrofias musculares de Duchenne y de Becker son enfermedades neuromusculares progresivas, con un patrón de herencia recesivo ligado al cromosoma X y causadas por mutaciones en el gen que codifica para la distrofina. El estudio de posibles portadoras en las familias afectadas resulta crucial, ya que genera expectativas y opciones frente al asesoramiento genético.
Objetivos: describir el diagnóstico molecular de distrofia muscular de Duchenne/Becker en una familia sin antecedentes patológicos de la enfermedad.
Métodos: se realizó un estudio experimental, de las deleciones en el gen distrofia muscular de Duchenne/Becker, en un paciente con diagnóstico clínico de la enfermedad, para lo cual se empleó la técnica de PCR-multiplex siguiendo los métodos descritos por Beggs y Chamberlain. También fueron estudiadas las mujeres de la familia, a través del análisis de marcadores polimórficos mediante repeticiones cortas en tándem de (CA)n.
Resultados: fueron identificadas en el paciente deleciones de los exones 47 al 52; así como la procedencia del cromosoma X ligado a la enfermedad (abuelo materno). Se determinó el estado de no portadora en tres mujeres de la familia. No se pudo excluir mosaicismo germinal en la madre del niño.
Conclusión: se infirió la ocurrencia de una mutación de novo. El diagnóstico molecular permitió la confirmación diagnóstica de la enfermedad en el niño afectado, además de la posibilidad de brindar un adecuado asesoramiento genético a la familia.
Palabras clave: distrofia muscular de duchenne, deleción cromosómica, patología molecular, enfermedades genéticas ligadas al cromosoma x.
ABSTRACT
Foundation: Duchenne and Becker muscular dystrophies are progressive neuromuscular diseases with a pattern of recessive inherited link to chromosome X and caused by mutations in the gene which codifies for dystrophin. The study of possible carriers in affected families is crucial since it generates expectations and options on genetic advisory.
Objective: to describe the molecular diagnosis of Duchenne/Becker muscular dystrophy in a family without pathological antecedents of the disease.
Methods: an experimental study was developed about the deletions of Duchenne/Becker gene of muscular dystrophy, in a patient with clinical diagnosis of the disease. It was used multiple PCR technique following the methods described by Beggs and Chamberlain. In addition, the women of the family were studied by the analysis of polymorphic markers through short repetitions in (CA) n tandem.
Results: deletions of exons from 47 to 52 were identified in the patient; so as the precedence of the X chromosome related to the disease (maternal grandfather). It was determined the state of non-carrier in three women of the family. It was not possible to exclude germline mosaicism in the child´s mother.
Conclusion: the occurrence of a novo mutation was inferred. The molecular diagnosis allowed confirming the diagnosis of the affected child; in addition it was possible to offer adequate genetic advisory to the family.
Key words: Muscular dystrophy, duchenne, chromosome deletion, pathology, molecular, genetic diseases, x-linked.
INTRODUCCIÓN
Las distrofias musculares de Duchenne (DMD) y de Becker (DMB) son enfermedades neuromusculares progresivas con un patrón de herencia ligado al cromosoma X, causadas por mutaciones (grandes deleciones, duplicaciones y pequeñas mutaciones) en el gen DMD que codifica para la proteína distrofina. Este gen contiene 2,4 Mb y está constituido por 79 exones.1-3 Las grandes deleciones de uno o más exones son las más frecuentes entre las mutaciones del gen y se presentan en el 68,5 % de los casos.1 El gen DMD presenta una alta tasa de mutación y se estima que uno de cada tres casos es causado por una mutación de novo.4,5 El modo de herencia recesivo ligado al cromosoma X, hace que las mujeres portadoras tengan un 50 % de probabilidad de tener hijos varones afectados e hijas portadoras. Esta información es crucial, ya que el conocimiento preciso de la condición de portadora o no portadora genera expectativas y opciones relacionadas con el asesoramiento genético.6-12 En Cuba, la introducción de algunas técnicas de biología molecular como la PCR (reacción en cadena de la polimerasa) -multiplex y el análisis indirecto mediante la construcción de los haplotipos usando repeticiones cortas en tándem de (CA)n (STR por su nombre en inglés Short Tandem Repeats), han hecho posible el análisis de este gen. Ello ha permitido la confirmación diagnóstica en niños con sintomatología clínica de DMD/B, así como la identificación de mujeres por línea materna portadoras de la enfermedad y el diagnóstico prenatal entre las semanas 18-20 del embarazo en aquellas familias con antecedentes de un niño afectado.13,14 El presente estudio tiene como objetivo describir el diagnóstico molecular de distrofia muscular de Duchenne/Becker en una familia sin antecedentes patológicos de la enfermedad.
MÉTODOS
Se realizó un estudio experimental para analizar las deleciones en el gen DMD en un paciente de tres años de edad, con diagnóstico clínico de la enfermedad; los marcadores polimórficos mediante repeticiones cortas en tándem de (CA)n en las mujeres de la familia para identificarlas como portadoras y no portadoras de la enfermedad; y el haplotipo en la familia. El niño comenzó a ser atendido en la consulta de neurogenética del Instituto de Neurología y Neurocirugía en el año 2012, debido a la presencia de sintomatología clínica de DMD. Luego de la confirmación diagnóstica, la familia asistió a la consulta de asesoramiento genético y se orientó entonces el estudio de las mujeres de la familia, para conocer su condición de portadoras. Previo consentimiento informado de padres del paciente y sujetos del estudio (seis miembros de la familia), se tomaron 5 ml de muestras de sangre periférica en tubos con EDTA al 5,6%, necesarios para el análisis indirecto. La extracción de ADN genómico se realizó mediante la técnica de precipitación salina.15 En el caso del paciente, la muestra se analizó para identificar las deleciones de 18 exones localizados en los puntos calientes del gen DMD mediante la técnica de PCR-multiplex según los métodos descritos previamente por Chamberlain y Beggs,16,17 con las variantes de tres reacciones independientes para el estudio de nueve exones siguiendo a Chamberlain (mezcla A: 8, 17, 44 y 51; mezcla B: 4, 12, 18 y 45; mezcla C: 44 y 48) y dos reacciones independientes para el estudio de los otros nueve exones siguiendo a Beggs (mezcla 6 exones: promotor, 3, 6, 13, 43, 52; mezcla 3 exones: 47, 50 y 60). El ADN de un sujeto masculino sano sirvió de control positivo, y una reacción sin ADN, como control negativo de amplificación. Posteriormente, 15 µL de los productos de amplificación y 5 µL del marcador de peso molecular pGEM (Promega) fueron resueltos en una electroforesis en gel de agarosa al 3 % y los geles, teñidos con bromuro de etidio. Los resultados fueron visualizados en un transiluminador (Bioblock Scientific). La condición de portadora o no portadora en las mujeres con riesgo se determinó a través del análisis de cuatro microsatélites o STRs situados en el gen DMD (3´CA, STR49, STR50 y 5´ DYS II), disponibles en el laboratorio en ese momento (Tabla 1) y la construcción de los haplotipos, mediante la identificación del alelo mutado por segregación de estos marcadores polimórficos.18,19 El haplotipo de cada STR fue establecido sobre la base de la movilidad electroforética de cada alelo. Para los STRs se usó una mezcla de PCR a un volumen final de 25 µL, con los reactivos y a las concentraciones siguientes: solución tampón STR 10X a 1X (Promega), 0,04 U/µl de Taq DNA polimerasa (Promega), 1,5 µM de cada primer Eurogentec y 100 ng de ADN genómico. Las amplificaciones se hicieron en un termociclador (Mini CyclerTM MJResearch, PTC-150) en las condiciones específicas para cada STR. Para los STRs 49 y 50, se hicieron a 94 ºC por cuatro minutos, 25 ciclos de 94 ºC por 30 segundos, 62 ºC por 30 segundos, 65 ºC por dos minutos y 65 ºC por siete minutos. Para el STR 3´CA se hicieron a 94 ºC por cinco minutos, 25 ciclos de 94 ºC por 15 segundos, 60 ºC por 30 segundos, 94 ºC por dos minutos y 60 ºC por cinco minutos. Para 5´DYSII se hicieron a 94 ºC por siete minutos, 25 ciclos de 94ºC por 30 segundos, 55 ºC por cuatro minutos, 72 ºC por un minuto y 72 ºC por diez minutos. Para cada estudio de STR se usó un control negativo de amplificación. Los productos amplificados (15 µl) y 5 µl de un marcador de peso molecular [pGEM (Promega) o Gel Pilot® DNA Molecular Weight Markers (QIAGEN)] se aplicaron sobre geles desnaturalizantes de poliacrilamida al 8 % y se corrieron en electroforesis vertical durante cuatro horas a 560 V, 50 mA. Posteriormente se tiñeron los geles en una solución de bromuro de etidio y se visualizaron en un transiluminador (Bioblock Scientific). En el año 2017 se adquirió en el laboratorio un fotodocumentador Multi Doc-It™ Imaging Systems UVP acoplado a un trasiluminador Benchtop UV UVP, y acorde a la disponibilidad de muestras de la familia, algunos STRs fueron repetidos y estos geles de poliacrilamida fueron retratados. Este trabajo ha sido aprobado por el Comité de Ética de la Investigación Científica y por el Consejo Científico del Instituto de Neurología y Neurocirugía.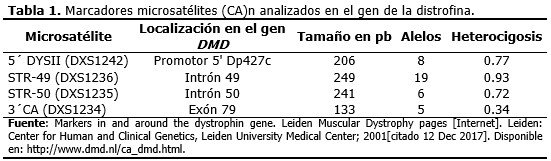
RESULTADOS
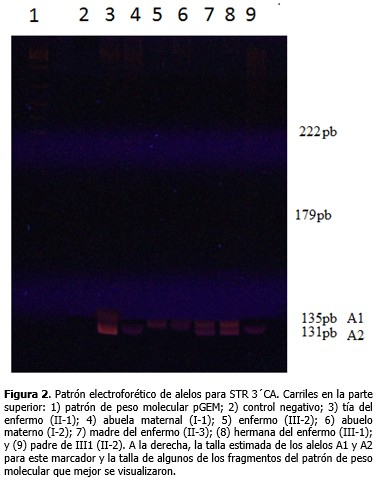
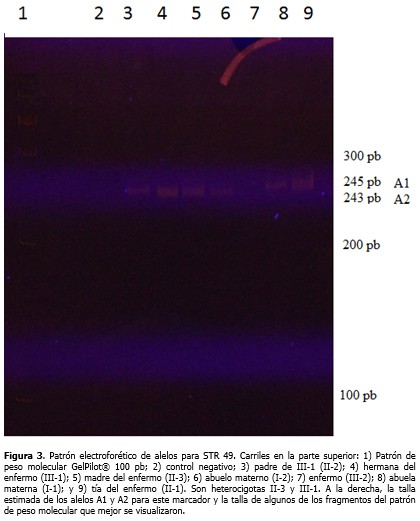
El estudio directo para los 18 exones evaluados del gen DMD permitió identificar en el paciente deleciones de los exones 47 al 52. Con los estudios indirectos se determinó el origen del haplotipo ligado a la enfermedad, gracias a la posibilidad de incluir en el análisis a la abuela y abuelo maternos. Los loci 3´CA, 5´ DYS II, STR 50 y STR 49 mostraron 2, 2, 3 y 2 alelos diferentes, respectivamente para esta familia. (Figura 1). La familia resultó informativa solo para los marcadores 3´CA y 5´DYS II, y con ellos se identificó que el cromosoma X portador de la deleción fue segregado a partir del abuelo materno (I-2), lo cual se observa más detalladamente a través del patrón electroforético de alelos para STR 3´CA. (Figura 2). Del análisis de haplotipos para estos marcadores, claramente se definió que la abuela (I-1) y tía del paciente (II-1) no eran portadoras y, por tanto, no presentaban riesgo de tener hijos afectados por DMD. Para los STRs 49 y 50 no se obtuvo amplificación en el paciente y para el STR 50 la madre del enfermo (II-3) mostró homocigocidad o pérdida de heterocigocidad. (Figura 1). La electroforesis del marcador STR 49 (Figura 3), evidenció no amplificación en carril del enfermo debido a la deleción que incluyó intrón 49 (deleción de los exones 47 al 52). Así mismo, se observó heterocigocidad en II-3, descartándose que la madre fuese una portadora obligada. Su hija (III-1) resultó no portadora dada su heterocigocidad para el locus 49 (Figura 3) y el 50 (Figura 1). 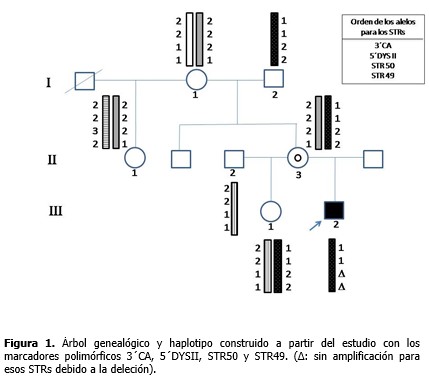
DISCUSIÓN
La no amplificación para los STRs 49 y 50 obtenida en el paciente es un resultado esperado, ya que estos marcadores polimórficos se localizan en la región del gen DMD que se encuentra mutado. La homocigocidad o pérdida de heterocigocidad obtenida para el STR 50 en la madre del enfermo, hizo pensar inicialmente que ella fuera una posible portadora de la deleción, producto de una mutación de novo ocurrida durante la espermatogénesis del abuelo materno. Sin embargo, la heterocigocidad obtenida en ella para el STR 49, descartó esa idea, sin dejar de lado un posible mosaicismo germinal. Los hallazgos de heterocigocidad obtenidos para los locus 49 y 50 en la hermana del niño condujeron a suponer una mutación de novo originada durante la ovogénesis materna de II-3 o durante la embriogénesis de III-2. El diagnóstico de mujeres portadoras de DMD/DMB ha sido un problema desconcertante por décadas. Las heterocigóticas son, por lo general, clínicamente normales y en casos raros (menos del 5 %) las portadoras muestran síntomas clínicos que pueden variar de moderados a severos. Por lo tanto, en una paciente adulta con debilidad muscular se debe considerar la determinación de su estado de portadora.20 Hoy en día la estrategia diagnóstica más seguida para esta enfermedad consiste en la identificación de deleciones y duplicaciones de exones mediante la técnica de amplificación múltiple de sondas dependientes de ligando, la cual permite examinar los 79 exones que posee el gen DMD. En caso de no hallarse estas mutaciones se procede a la secuenciación de las regiones codificantes y sitios de empalme para la búsqueda de mutaciones puntuales.21-30 Estas mismas técnicas pueden ser utilizadas para la detección de las mutaciones en mujeres en riesgo, pero se recomienda que siempre que sea posible se identifique la mutación en el caso índice o en una portadora obligada de la familia. De otro modo, un resultado negativo es difícil de interpretar, si se considera que estos métodos no son totalmente sensibles y tienen un riesgo residual aproximado al 3 %, aun después de combinadas las técnicas.18 Otras técnicas también han sido utilizadas, tales como: southern blot, hibridación fluorescente in situ y determinación de dosis génica con densitometría o mediante PCR en tiempo real.6,7,21-30 En el laboratorio donde se realizó el estudio, estas técnicas aún no están disponibles. Solo se estudian las deleciones de 18 exones de los 79 exones que posee el gen DMD mediante PCR multiplex y se emplean los polimorfismos (CA)n localizados en este, útiles para la detección de portadoras, mosaicismos germinales y mutaciones de novo.31-33 Según la literatura, las mutaciones de novo de tipo puntual y duplicaciones, surgen preferiblemente durante la espermatogésis, mientras que deleciones ocurren principalmente durante la ovogénisis;20 y en los casos esporádicos son más frecuentes las deleciones distales.34 Grimm y colaboradores20 describen para la DMD/DMB tres situaciones en las que se hereda una mutación en el gen DMD: 1) la madre de un hijo afectado es portadora porque su madre es portadora; 2) la madre de un hijo afectado es portadora como resultado de una mutación de novo que ha surgido en la meiosis durante la espermatogénesis del abuelo, la ovogénesis de la abuela o la ovogénesis de la madre y 3) la madre de un hijo afectado es portadora como resultado de una mutación de novo que ha surgido en la mitosis con la consecuencia de un mosaicismo germinal, durante la espermatogénesis del abuelo, la ovogénesis de la abuela o la ovogénesis de la madre. En la madre de este niño no se pudo descartar la existencia de un mosaicismo germinal, lo que conllevaría un alto riesgo de ser portadora, aunque pudiera no serlo debido a la ocurrencia de una mutación meiótica de novo en el probando durante la embriogénesis. Para un caso esporádico donde se confirme una deleción se estima que esa madre tiene la probabilidad de 0,247 de presentar un mosaicismo germinal y de 0,173 de que sea consecuencia de una mutación de novo durante la división meiótica en el enfermo.20 El riesgo de recurrencia (riesgo de un segundo hijo varón afectado) para mujeres no portadoras debido a mosaicismo germinal ha sido estimado entre 14-20 % si el haplotipo de riesgo es trasmitido. Helderman y colaboradores estimaron que en familias con un caso de DMD esporádico de mutación desconocida, el riesgo de recurrencia es más bajo (8,6 %), e incluso, de 4,3 % cuando no existe información sobre el haplotipo de riesgo. Este riesgo es diferente en dependencia del tipo de mutación –deleciones proximales, deleciones distales, duplicaciones y mutaciones puntuales–.35 De cualquier modo, en estos casos, para descartar el riesgo de tener otro hijo afectado se recomienda el diagnóstico prenatal.20 Es significativo recalcar que en los casos DMD/DMB esporádicos con diagnóstico molecular confirmado es importante realizar los estudios por STRs que involucren a varios miembros de la familia, incluyendo los abuelos maternos, pues ello permite identificar correctamente el origen del alelo mutado. Hasta la fecha se conoce de 419 familias con sospecha clínica de la enfermedad, documentadas en el Libro de Registro Nacional de DMD/DMB, y de ellas solo 121 han solicitado estudios de mujeres portadoras, el 57 % (69/121) correspondiente a casos esporádicos y el 43 % (52/121) a casos familiares. Estas cifras alertan sobre la necesidad de ofrecer un adecuado asesoramiento genético a estas familias, para lograr identificar los riesgos y disminuir la incidencia de la enfermedad. Puede concluirse que los estudios moleculares realizados permitieron la confirmación diagnóstica de DMD/B en el paciente mediante la identificación de la deleción de los exones 47 al 52, así como la identificación del estado de no portadoras en tres de las cuatro mujeres de la familia estudiadas. Se sugiere la ocurrencia de una mutación de novo. No se pudo excluir un mosaicismo germinal en la madre del paciente. Con estos elementos se pudo brindar un adecuado asesoramiento genético a la familia, al ofrecer una mayor explicación de los riesgos y las opciones reproductivas.
REFERENCIAS BIBLIOGRÁFICAS
1. Bladen C, Salgado D, Monges S, Foncuberta ME, Kekou K, KosmaK, et al. The TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy Mutations. Hum Mutat. 2015;36(4):395-402
2. Le Rumeur E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn J Basic Med Sci. 2015;15(3):14-20
3. Vieitez I, Gallano P, González L, Borrego S, Marcos I, Millán JM, et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: Study of 284 cases. Neurologia. 2017;32(6):377-85
4. Laing NG. Molecular genetics and genetic counselling for Duchenne/Becker muscular dystrophy. Mol Cell Biol Hum Dis Ser. 1993;3:37-84
5. Garcia S, de Haro T, Zafra-Ceres M, Poyatos A, Gomez-Capilla JA, Gomez-Llorente C. Identification of de novo mutations of Duchénnè/Becker muscular dystrophies in southern Spain. Int J Med Sci. 2014;11(10):988-93
6. Delgado WN, Borjas L, Zabala W, Fernández E, Sóliz E, Chavez C, et al. Detección de portadoras de distrofia muscular de Duchenne/Becker a través del análisis de los loci STRs ligados al gen de la distrofina en familias venezolanas. Invest Clin. 2002;43:239-54
7. Fonseca D, Tamar C, Mateus H. Detección de portadoras de Distrofia Muscular de Duchenne en familias colombianas mediante análisis de microsatélites. Colombia Médica. 2008;39 Suppl 2:7-13
8. Vears DF, Delany C, Massie J, Gillam L. Why Do Parents Want to Know their Child’s Carrier Status? A Qualitative Study. J Genet Couns. 2016;25(5):1257-66
9. Stark AE. Determinants of the incidence of Duchenne muscular dystrophy. Ann Transl Med. 2015;3(19):287
10. Bogue L, Peay H, Martin A, Lucas A, Ramchandren S. Knowledge of carrier status and barriers to testing among mothers of sons with Duchenne or Becker muscular dystrophy. Neuromuscul Disord. 2016;16(12):860-4
11. Hayes B, Hassed S, Chaloner JL, Aston CE, Guy C. Duchenne Muscular Dystrophy: a Survey of Perspectives on Carrier Testing and Communication Within the Family. J Genet Couns. 2016;25(3):443-53
12. Helderman-van den Enden AT, Madan K, Breuning MH, van der Hout AH, Bakker E, de Die-Smulders CE, et al. An urgent need for a change in policy revealed by a study on prenatal testing for Duchenne muscular dystrophy. Eur J Hum Genet. 2013;21(1):21-6
13. Perera H, Zaldivar T, Viñas C, Medina A, Guerra R, Zayas M. Diagnóstico prenatal de la distrofia muscular de Duchenne. Presentación de una familia. Medisur [revista en Internet]. 2007 [citado 1 Ago 2018];5(1):[aprox. 6p]. Disponible en: http://www.medisur.sld.cu/index.php/medisur/article/view/247/3710
14. Rodríguez M, Ferreira R, Gayol LA, Quintana J, Rendón R, Méndez D. Diagnóstico de distrofia muscular de Duchenne mediante análisis del ácido desoxinucleótico y su aplicación en la prevención. Rev Cubana Pediatr [revista en Internet]. 1996 [citado 1 Ago 2018];68(1):[aprox. 12p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75311996000100003&lng=es
15. Miller S, Dykes D, Polesky H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988;16(3):1215
16. Beggs AH, Koening M, Boyce FM, Kunkel LM. Detection of 98 % of DMD/DMBMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86(1):45-48
17. Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16(23):11141-56
18. Clemens PR, Fenwick RG, Chamberlain JS, Gibbs RA, de Andrade M, Chakraborty R, et al. Carrier detection and prenatal diagnosis in Duchenne and Becker muscular dystrophy families, using dinucleotide repeat polymorphisms. Am J Hum Genet. 1991;49(5):951-60
19. Feener CA, Boyce FM, Kunkel LM. Rapid detection of CA polymorphisms in cloned DNA: application to the 5’ region of the dystrophin gene. Am J Hum Genet. 1991;48(3):621-7
20. Grimm T, Kress W, Meng G, Müller CR. Risk assessment and genetic counseling in families with Duchenne muscular dystrophy. Acta Myol. 2012;31(3):179-83
21. Juan-Mateu J, Gallano, Trujillo-Tiebas MJ; Grupo AEGH/CIBERE. Recomendaciones de buena práctica para el diagnóstico genético de las distrofias musculares de Duchenne y de Becker. Med Clin (Barc). 2012;139(7):307-12
22. Okubo M, Minami N, Goto K, Goto Y, Noguchi S, Mitsuhashi S, et al. Genetic diagnosis of Duchenne/Becker muscular dystrophy using next-generation sequencing: validation analysis of DMD mutations. J Hum Genet. 2016;61(6):483-9
23. Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53(3):145-51
24. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, CripeL, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77-93
25. Nascimento A, Medina J, Camacho A, Madruga M, Vilchez JJ. Consenso para el diagnóstico, tratamiento y seguimiento del paciente con distrofia muscular de Duchenne. Neurología [revista en Internet]. 2018 [citado 10 Sep 2018];33(8):[aprox. 20p]. Disponible en: https://www.sciencedirect.com/science/article/pii/S021348531830015X?via=ihub
26. Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251-67
27. Araujo APQC, Carvalho AAS, Cavalcanti EBU, Saute JAM, Carvalho E, França MC, et al. Brazilian consensus on Duchenne muscular dystrophy. Part 1: diagnosis, steroid therapy and perspectives. Arq Neuropsiquiatr. 2017;75(8):104-13
28. Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT. Mutational Spectrum of DMD Mutations in Dystrophinopathy Patients: Application of Modern Diagnostic Techniques to a Large Cohort. Hum Mutat. 2009;30(12):1657-66
29. Suh MR, Lee KA, Kim EY, Jung JH, Choi WA, Kang SW. Multiplex Ligation-Dependent Probe Amplification in X-linked Recessive Muscular Dystrophy in Korean Subjects. Yonsei Med J . 2017;58(3):613-8
30. Bello L, Pegorano E. Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy. Acta Myol. 2016;35(3):122-7
31. Chaturvedi LS, Mittal RD, Srivastava S, Mukherjee M, Mittal B. Analysis of dinucleotide repeat loci of dystrophin gene for carrier detection, germline mosaicism and de novo mutations in Duchenne/Becker muscular dystrophy. Clin Genet. 2000;58(3):234-5
32. Fonseca DJ, Mateus H, Silva CT. Pérdida de heterocigocidad e identificación de portadoras de distrofia muscular de Duchenne: un caso familiar con evento de recombinación. Rev Cienc Salud. 2012;10(1):83-90
33. Mukherjee M, Chaturvedi LS, Srivastava S, Mittal RD, Mittal B. De novo mutations in sporadic deletional Duchenne muscular dystrophy (DMD) cases. Exp Mol Med. 2003;35(2):113-7
34. Passos-Bueno MR, Bakker E, Kneppers AL, Takata RI, Rapaport D, den Dunnen JT, et al. Different mosaicism frequencies for proximal and distal Duchenne muscular dystrophy (DMD) mutations indicate difference in etiology and recurrence risk. Am J Hum Genet. 1992;51:1150-5
35. Helderman-van den Enden AT, de Jong R, den Dunnen JT, Houwing JJ, Kneppers AL, Ginjaara HB, et al. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clin Genet. 2009;75(5):465-72
Recibido: 19 de diciembre de 2017.
Aprobado: 18 de octubre de 2018.
Ivonne Martín Hernández. Licenciada en Bioquímica. Máster en Bioética. Investigador Auxiliar. Profesor Auxiliar Correo electrónico: ivonne.martin@infomed.sld.cu

