










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La hipoglucemia hiperinsulinémica endógena persistente es causada por la alteración en la función de la célula β del páncreas. Suele presentarse en dos situaciones patológicas del páncreas donde se secreta excesiva cantidad de insulina: la más frecuente es el insulinoma, presentada en los adultos; y menos común, la hiperplasia de las células de los islotes de Langerhans, conocida como nesidioblatosis o síndrome hipoglucémico pancreático no insulinoma.1
La nesidioblastosis fue descrita por primera vez en 1938 por George Laidlaw. quien la definió como un trastorno raro que resulta de la neoformación de células de los islotes de Langerhans en el epitelio ductal pancreático exocrino.2
Actualmente la nesidioblastosis del adulto se define como cambios en el páncreas endocrino, caracterizados por la proliferación anormal de las células de los islotes pancreáticos, que afecta en forma difusa a la glándula. El sustrato morfológico de estos cambios moleculares puede presentarse en los niños, como hipertrofia difusa de la célula β (nesidioblastosis difusa) en el 60 %, o hipertrofia focal de la célula β (nesidioblastosis focal) en el 40 %; esta última forma no ha sido descrita en adultos.3
El síndrome de neoplasia endocrina múltiple de tipo 1 (MEN 1) o síndrome Werner, fue descrito por primera vez en 1954 por Paul Wermer, quien indicó que el MEN 1 está causado por mutaciones germinales inactivadoras en heterocigosis en el gen MEN 1 del cromosoma 11 (11q13), el cual codifica la proteína menina, un supresor tumoral.4,5
El síndrome de Wermer o síndrome MEN 1 es un trastorno hereditario de tipo autosómico dominante, con una incidencia de 1 a 10 por cada 100 000 personas, donde el 90 % de los pacientes presentan un familiar de primer orden afectado por esta entidad, y solo el 10 % presenta mutaciones de novo.6,7,8 Se caracteriza por la presentación en el mismo sujeto de la afectación de una o más glándulas (endocrinas o no), que pueden ser tumores de paratiroides, hipófisis y duodenopancreáticos.9
En pacientes con un único tumor y sin historia familiar, la probabilidad de que exista mutación en MEN 1 es mayor cuanto menor es la edad de diagnóstico, o si existen múltiples tumores en el órgano afectado. El tratamiento de la nesidioblastosis, al igual que el insulinoma, es la resección quirúrgica del páncreas, y es controversial la extensión de la cirugía.10 La cirugía raramente es curativa, y precisa tratamiento médico asociando al diazóxido, octreótido e interferón alfa.
Se presenta el caso. Debido a la poca frecuencia de esta enfermedad, resulta de interés describir el caso de un paciente adulto con cuadros recurrentes de hipoglucemia secundarios a hiperinsulinismo endógeno, con el objetivo de exponer las principales manifestaciones clínicas y conducta a seguir.
PRESENTACIÓN DEL CASO
Se presenta el caso de un paciente masculino, de 23 años, con antecedentes de salud de nesidioblastosis diagnosticado a los 16 años de edad. Inició su padecimiento a los 14 años, con episodios de convulsiones; para esto recibió medicamento anticonvulsivante por más de seis años. Se sumaron al cuadro clínico episodios repetidos de hipoglucemia, por lo cual requirió hospitalizaciones para manejo únicamente con solución glucosada. Fue ingresado para mejor estudio y tratamiento del cuadro. Tras la realización de analíticas y ecografía pancreático endoscópica, se concluyó páncreas compatible con nesidioblastosis. No se contó con diagnostico histopatológico. Desde ese momento fue tratado con diazóxido y alimentación fraccionada seis veces al día de forma indefinida. No acudió a seguimiento ni control médico por espacio de dos años.
Fue recibido en el Hospital General Docente Ambato por presentar convulsiones tónico clónicas, hipoglucemia severa (glucosa 13mg/dl) con posterior estatus epiléptico, para lo cual requirió manejo avanzado por vía aérea con ventilación mecánica durante tres días. Presentó mejoría tras la administración de soluciones glucosadas, dextrosa al 50 % intravenoso a 60 ml/hora, consiguiendo control glucémico sobre los 90 mg/dl.
El paciente refirió haber presentado marcadas hipoglucemias (44 mg/dl glucemia capilar) de forma reiterada en la última semana, caracterizadas por diaforesis, palpitaciones y malestar general; aumento de peso de aproximadamente 40 kg en dos años, por exceso de calorías consumidas para poder evitar cuadro de hipoglucemia.
Al examen físico se determinó: talla de 1,75 m, peso de 150 kg, con obesidad mórbida (IMC=49 kg/m2); cifras de tensión arterial de 120/69 mmHg; frecuencia cardiaca de 105 latidos por minuto. Se constató acantosis nigricans marcada en cuello, axilas y región inguinal, con presencia de acrocordones en cuello y axilas. El resto del examen físico no identificó datos de interés.
Se realizó estudio hormonal, que mostró en test de ayuno valores de insulina = 109 mUI/ml (valor de referencia: 6 - 24); Péptido C = 13,7 ng/ ml (valor de referencia: 1,1-4,4). Estos resultados confirmaron el diagnóstico bioquímico de hipoglucemia hiperinsulinémica.
Por persistencia de sintomatología durante su internamiento fue necesario mantener infusión glucosada (dextrosa al 50 %) permanentemente a 80 ml/hora. Se decidió trasferencia a hospital de mayor complejidad para resolución quirúrgica.
En su estancia hospitalaria se profundizó en estudios de imagen. La ecografía de cuello mostró tiroides de tamaño conservado, simétrica, con volumen adecuado; llamó la atención a nivel infratiroideo derecho imagen hipoecogénica con vascularidad periférica presente, de aproximadamente 21x27 mm en relación probable con adenoma paratiroideo.
La tomografía axial computarizada simple y contrastada de abdomen con protocolo pancreático mostró lesión de aproximadamente 4x3,4 cm, dependiente de cabeza de páncreas, sugerente según clínica del paciente de insulinoma (Fig. 1). No se pudo realizar resonancia magnética simple y contrastada de hipófisis debido a dimensiones corporales del paciente que imposibilitaron acceso al resonador.
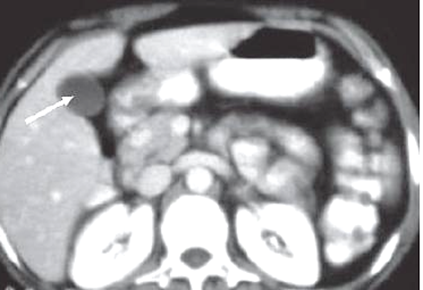
Fig. 1 Tomografía axial computarizada simple y contrastada que mostró lesión de aproximadamente 4x3,4 cm, dependiente de cabeza de páncreas.
Algunos valores de interés derivados del estudio hormonal fueron paratohormona (pth): 650,7; calcio: 11; hormona estimulante de la tiroides (TSH): 3,48; prolactina: 45,77; y testosterona total: 2,910.
Tras analizar el caso, se planteó la presencia de dos criterios de neoplasia endocrina múltiple tipo 1: insulinoma e hiperparatiroidismo primario. De manera que se redefinió el diagnóstico inicial. Fue valorado por el servicio de Genética, para la realización del diagnóstico molecular al paciente y a otros integrantes de la familia (diagnóstico molecular precoz).
Se indicó tratamiento con bifosfonato (ácido zoledrónico 4 miligramos) y se planificó interconsulta con el servicio de Cirugía General, para programación de resolución quirúrgica acorde al diagnóstico.
DISCUSIÓN
La mayoría de los pacientes con hipoglucemia por hiperinsulinismo presentan los síntomas típicos de hipoglucemia, síntomas adrenérgicos como diaforesis, palpitaciones, ansiedad, temblor, sensación de hambre, y síntomas de neuroglucopenia como confusión, visión borrosa, amnesia y pérdida de la conciencia, valores plasmáticos bajos de glucosa y alivio de dichos síntomas al aumentar la concentración plasmática de glucosa.11) Varios de estos síntomas fueron identificados en el paciente referido en el presente caso.
La distinción preoperatoria entre nesidioblastosis e insulinoma puede ser difícil, ya que la presentación bioquímica de ambas es similar, y los estudios de imagen son a menudo equívocos.3
Clínica, bioquímicamente y por estudio de imagen resulta difícil distinguir entre la nesidioblastosis difusa y el insulinoma. Por tanto, al profundizar el estudio y nuevamente reevaluar elementos endocrinos se definió como una neoplasia endocrina múltiple tipo 1.
MEN 1 es una enfermedad caracterizada por la aparición de hiperfunción o hiperplasia de dos o más glándulas endocrinas y la aparición de tumores glandulares, generalmente benignos, en glándulas la paratiroides, hipófisis y duodenopancreáticos. Entre el 30 y el 80 % de los portadores de mutación de MEN 1 desarrollan tumores neuroendocrinos duodenopancreáticas (TNEP), que pueden empezar a manifestarse a partir de la adolescencia. Las neoplasias descritas con mayor frecuencia son gastrinoma, insulinoma, glucoganoma, carcinoide gástrico y somatostinoma; en un 30-40 % de los casos pueden desarrollarse tumores no funcionantes.12 De igual forma se describen otras manifestaciones como los colagenomas.13
La clínica del síndrome MEN 1 suele ser variable de unos sujetos a otros, tanto en el número de órganos afectos, como en la cronología del inicio de las diferentes afecciones y la forma de presentación de la enfermedad; ello hace que el diagnóstico basado en la clínica suela ser difícil y generalmente tardío. Aunque inicialmente la sintomatología es leve y la afectación glandular se presenta como hiperplasia, si se deja evolucionar la enfermedad sin tratamiento hasta el 48 % de los pacientes con MEN 1 pueden fallecer por causas secundarias a la enfermedad, de ahí la importancia de un diagnóstico precoz.14,15,16
El hiperparatiroidismo primario es la enfermedad endocrina más prevalente en el síndrome MEN 1 y afecta prácticamente al 100 % de los pacientes con este proceso.9
Según exponen Torres y colaboradores,2 la tomografía computada multicorte, la resonancia magnética y el ultrasonido endoscópico permiten una correlación directa entre el tumor y el resto del parénquima pancreático. Al ser estudios no invasivos, pueden ser considerados de primera línea para la caracterización morfológica de los insulinomas. Similares consideraciones son expuestas por otros expertos(15, 16) y consensos.17
En el presente caso se detectaron, mediante tomografía axial computarizada, imágenes sugerentes de un tumor en cabeza de páncreas compatible con insulinoma; y de adenoma paratiroideo mediante ecografía de cuello.
El tratamiento de la nesidioblastosis llevado por el paciente por más de seis años mediante la incorporación de alimentos fraccionados en varias ingestas diarias para evitar la hipoglucemia y en crisis de esta, desarrolló obesidad mórbida y signos clínicos de resistencia a la insulina, cursando con glucosa alterada en ayunas. En el último ingreso, tras la valoración médica y concluyente con cuadro de MEN tipo 1 se decidió programación para resección quirúrgica del páncreas y estudio genético para familiares.
El cribado de las mutaciones de MEN 1 tiene varias ventajas, entre las que se incluyen la confirmación del diagnóstico clínico y la identificación de los miembros de la familia portadores de mutaciones de NEM 1, lo cual permite adoptar las alternativas terapéuticas y el seguimiento adecuado. Actualmente, las pruebas genéticas para detectar defectos en el gen MEN1 incluyen el cribado mediante PCR de mutaciones en la región codificante y en las uniones de empalme. Si no se identifica una mutación mediante el método mencionado, se realiza un cribado basado en la amplificación de sondas de ligación múltiple (MLPA) para detectar grandes deleciones del gen MEN 1.6
Los pacientes con MEN 1, presentan una reducción de la esperanza de vida frente a los sujetos normales, de tal forma que, el diagnóstico precoz del síndrome mediante el estudio genético y el posterior diagnóstico y tratamiento de las del MEN 1 en fases iniciales permite tasas de curación superiores. En un estudio realizado por Beek y colaboradores,10 se les realizó una resección por insulinoma en el contexto del MEN 1 a 96 pacientes; 66 presentaron insulinoma localizado y 33 insulinoma multifocal. Tras un seguimiento de 7,8 años, un paciente presentó enfermedad persistente y seis enfermedades recidivantes, y cuatro, un nuevo insulinoma. A los 10 años se presentó una supervivencia libre de hipoglucemia del 91 %.
La hipoglucemia hiperinsulinémica endógena constituye un reto para la Medicina; el diagnóstico de su causa en cada paciente constituye una prioridad. Ante pacientes afectados por esta entidad, secundaria a una tumoración, o cualquier paciente con un tumor de glándulas endocrinas, debe descartarse la presencia de un síndrome de neoplasia endocrina múltiple de tipo 1. La definición molecular permite el diagnóstico y asesoramiento genético a todos los integrantes de la familia en cuanto a la posibilidad de presentar la mutación y desarrollar la enfermedad, así como de transmitirla a su descendencia.

