










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
versión On-line ISSN 1729-519X
Rev haban cienc méd v.9 n.2 Ciudad de La Habana abr.-jun. 2010
CIENCIAS CLÍNICAS Y PATOLÓGICAS
Hospital Neumológico Benéfico Jurídico
Fibrosis quística. Diagnóstico tardío en el adulto presentación de caso
Delayed diagnosis of the cystic fibrosis in adulthood. A case report
Mireya Fernández Fernández1, Alfredo Jané Lara2, Fidel Rodríguez Cala3, Hilda García Castañeda4, Sergio Fernández García5, Hilda Roblejo Balbuena6
1Especialista Primer Grado en Neumología. 280 entre 243 y 243A número 24323 apto 3. Reparto INAV. Wajay. Boyeros. mireyaff@infomed.sld.cu 2Especialista Primer Grado en Medicina Interna y Segundo Grado en Neumología. Auxiliar. Calle 17 núm. 1369. El Vedado. Municipio Plaza. Ciudad de La Habana.ajane@infomed.sld.cu
3Especialista Segundo Grado en Neumología. Cristo núm.27 entre Muralla y Teniente Rey .Habana Vieja.Ciudad de La Habana.Teléfono:8327151. 4Especialista Primer Grado en Neumología. Calle 208 entre 33 y 37 edificio 32. Apartamento 18. La Lisa. Ciudad de La Habana.
5Especialista Primer Grado en MGI y Neumología. Instructor. Amarera 104 entre Venus y Bertemati Guanabacoa. Ciudad de La Habana.
6Especialista Primer Grado en MGI y Genética Clínica. San Nicolás 220 apto 6 entre Concordia y Virtudes. Centro Habana. Ciudad de La Habana.
RESUMEN
Se presenta el caso de un paciente blanco de 47 años con cuadros respiratorios a repetición que cedían con tratamiento sintomático o con el uso de antibióticos por vía oral desde la infancia. A los 39 años de edad, comenzó con agudizaciones más frecuentes que requirieron tratamientos con antibióticos e ingresos en varios hospitales de Ciudad de La Habana, y se le diagnosticó Bronquiectasias diseminadas bilaterales e infectadas con Pseudomonas Aeruginosas, secundarias a una posible Tuberculosis adquirida en la antigua URSS a los 21 años. La falta de lesiones hacia los vértices pulmonares en la radiografía de tórax y el hecho de no haber nunca tenido hijos despertó la sospecha clínica de Fibrosis Quística, la cual fue confirmada por los estudios realizados posteriormente.
Nuestro propósito al presentar este caso es alertar que esta enfermedad ha dejado de ser una patología exclusiva de Pediatría y que debe sospecharse ante un cuadro florido de bronquiectasias.
Palabras clave: Bronquiectasias, Pseudomonas, Fibrosis Quística.
ABSTRACT
The case of a 47 years old white patient, that presented repeated respiratory symptoms that only improve using symptomatic treatment or oral antibiotics. In 2000 when, he was 39 years old their symptoms increased and he required antibiotic intensive treatment and incomes at several hospital in Havana.
After that, he was diagnosed with multiple bronchiectasis spread bilateral and infected Pseudomona Auriginosa, that could brings about, TB acquired at URSS when he was 21 years old.
In 2008 because of the absence of lesions toward pulmonary apex in the Rx and the fact he couldn't have children aroused the clinical suspicion of Cystic Fibrosis and it was later confirmed by the accomplished studies.
Our purpose to present this case is to alert and prevent that this disease has stopped being Pediatric's exclusive pathology and it should be taking into account when there is a complete picture of a multiple spread bronchiectasis.
Key words : Bronchiectasis, Pseudomonas, Cystic Fibrosis.
INTRODUCCIÓN
La Fibrosis Quística (FQ) es una enfermedad monogénica con patrón de herencia autosómico recesivo. Su incidencia varía de 1 entre 3 000 a 1 entre 8 000 nacidos vivos.1
En 1936, Fanconi et al 2 hicieron la primera descripción de la FQ y asociaron la fibrosis congénita del páncreas con la presencia de bronquiectasias. En 1934, Andersen 3 describe los hallazgos clínicos y anatomopatológicos de los enfermos con FQ, y denominó por primera vez con el nombre de fibrosis quística del páncreas a esta enfermedad, por los quistes y fibrosis del tejido pancreático que presentan estos enfermos.
En 1985, se identificó y localizó el locus responsable de la FQ en 7q31-32, 4 y cuatro años más tarde, se comunicó el descubrimiento del gen responsable de la enfermedad, 5-6-7 que codifica una proteína reguladora de la conductancia transmembrana: cystic fibrosis transmembrane conductance regulador (CFTR), y demostró al mismo tiempo que la mutación más común es la DF508, aunque hoy se sabe que existen más de mil mutaciones para este gen. 8 La naturaleza de las mutaciones se correlaciona con la gravedad del cuadro clínico. La proteína CFTR se localiza en la membrana apical de muchos tipos de células epiteliales y el resultado final de todas las mutaciones detectadas que alteran la función de CFTR es el mismo: la imposibilidad de transportar cloruro. Esto explica la historia natural de la enfermedad en las glándulas sudoríparas, aparato respiratorio, páncreas, aparato genital masculino y el sistema hepatobiliar.1
Los pacientes con cuadros clásicos de la enfermedad suelen desarrollar, desde los primeros meses de vida, manifestaciones clínicas digestivas y/o respiratorias de la enfermedad. Además, existe un grupo de pacientes, quienes pueden cursar con manifestaciones clínicas leves o atípicas de la enfermedad durante los primeros años de la vida y que son diagnosticados en la edad adulta.9
Presentación del caso
Paciente masculino de 47 años de edad, de la raza blanca, con antecedentes desde la infancia de presentar Asma Bronquial y Rinitis Alérgica, asociada a los cambios climáticos, olores fuertes y el polvo; presentaba catarros frecuentes. En dos ocasiones, tuvo procesos infecciosos pulmonares asociados a esputos hemoptoicos. A los 21 años de edad, presentó Tuberculosis pulmonar en la URSS, para la cual llevó tratamiento específico. A los 34 años de edad, comienza nuevamente seguimiento por la especialidad de Alergia por el agravamiento de la Rinitis y se le diagnostica Sinusitis. Durante todo este tiempo, el paciente presentaba infecciones respiratorias frecuentes que cedían con tratamiento sintomático o con el uso de antibióticos por vía oral, y no requerían ingresos. En el año 2000, a los 39 años, comenzó con procesos infecciosos pulmonares muy frecuentes que necesitaban tratamiento con antibióticos sistémicos; se le diagnosticó Bronquiectasias múltiples bilaterales y extensas secundarias a la tuberculosis. Es valorado por nosotros en el 2008, presenta tos y expectoración purulenta diaria, disnea asociada a las exacerbaciones infecciosas y a los grandes esfuerzos. Presenta además síntomas dispépticos por una colecistopatía calculosa.
Examen Físico: Peso: 70Kg, Talla: 167cm.
Auscultación del Aparato Respiratorio: Murmullo vesicular disminuido globalmente, estertores roncos aislados en ambos campos pulmonares y subcrepitantes hacia las bases. No tiene otros datos positivos al exámen físico.
Teniendo en cuenta el antecedente de procesos respiratorios infecciosos frecuentes desde la infancia y observando que las lesiones que el paciente presentaba en la radiografía de tórax (Bronquiectasias) no eran sugestivas de ser secundarias a una Tuberculosis pulmonar y tener el antecedente de una infertilidad nunca antes estudiada, comenzamos el estudio por la sospecha de una Fibrosis Quística.
Se realizaron los siguientes estudios:
Hemograma con diferencial: Hb 13.9 gm% leuco 6,000 mm3, diferencial: normal. Eritrosedimentación: 45 mm/h.
Estudio inmunológico: IgA 2.05 g/l, IgM 0.71g/l, IgG 6.48g/l, IgE 180.93g/l. Estudio de la función hepática: TGP 20.7 U/I, TGP 10.6 U/I, GGT 31.0 U/L, LAP 31.40U/L, Fosfatasa Alcalina 157U/L.
Ultrasonido Abdominal: Litiasis vesicular.
Espermograma: Azoospermia. glicemia 4.30mmol/l, creatinina 99mmol/l, conteo de eosinófilos 285mm3.
Electrolitos en sudor: Primera prueba: Cl - 180.0 mmol/l. Sudó 157mg. Segunda prueba: Cl _ 101.4 mmol/l. Sudó 287mg.
Estudio genético: No presenta las mutaciones: F508del, G542X, R1162X, R334W, R553X, 3120+1G>A, analizadas por métodos directos de biología molecular.
Estudio bacteriológico y BAAR del esputo y lavado bronquial: El crecimiento bacteriano obtenido fue de Pseudomona Aeruginosa variedad mucoide. En una exacerbación se aisló Estafilococo Aureus. El examen de las muestras para BAAR directo y cultivo fue negativo.
Estudio Funcional Respiratorio: Disfunción ventilatoria de tipo obstructiva pura de moderada intensidad irreversible al aerosol broncodilatador. Volúmenes pulmonares por termodilución del gas Helio (TLC, RV, FRC) y Capacidad de Difusión del Monóxido de Carbono con multigás de monóxido de carbono, metano, oxígeno, balanceado con nitrógeno (DLCO, KCO): Normales. Fibrobroncoscopía: Tráquea y Carina sin alteración.
Arbol bronquial derecho: Abundantes secreciones purulentas blanca-amarillenta como alteración del contenido, emergiendo más del lóbulo superior derecho.
Arbol bronquial izquierdo: Escasas secreciones purulentas.
Imagenología: Rx de tórax vista PA y Lateral: Opacidades difusas, de aspecto quístico en ambos campos pulmonares, mayores en el lóbulo superior derecho. No lesiones en vértices. Rx de senos perinasales: Opacidad del seno frontal derecho así como de ambos senos maxilares, más marcado en el izquierdo. Engrosamiento mucoso de los cornetes nasales.
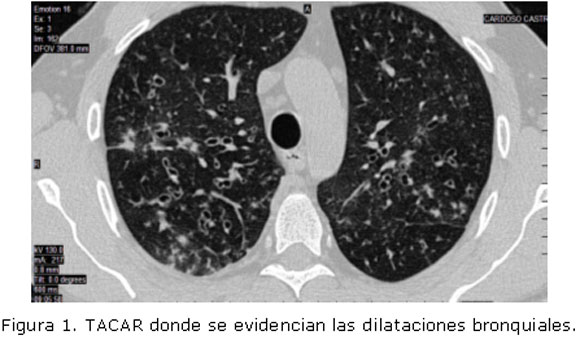
Tomografía axial computarizada de alta resolución (TACAR): Bronquiectasias quísticas y saculares en ambos campos pulmonares. (Figura 1).
DISCUSIÓN
La reunión de consenso sobre diagnóstico de la FQ, promovida por la Fundación Americana de Fibrosis Quística, acordó que los criterios diagnósticos actuales incluyen uno o más rasgos fenotípicos característicos o historia de Fibrosis Quística en hermano o primo hermano o pesquisaje neonatal positivo, más evidencia de disfunción del CFTR demostrada por dos determinaciones de concentraciones de cloro en sudor superior a 60 mmol/l, o demostración de alteraciones en el transporte iónico a través del epitelio nasal (diferencia de potencial nasal mayor de 35 mega voltiusl) o la detección de dos mutaciones reconocidas de (FQ).1
La presentación clínica de la enfermedad, la gravedad y su ritmo de progresión varía considerablemente, algunas de las variaciones podrían estar relacionadas con las diferentes mutaciones del gen de la Fibrosis Quística. Se han descrito 5 mecanismos moleculares, cuyo efecto predice una alteración o disfunción del CFTR. La mayoría de las mutaciones que no produce CFTR (clase I) o que la proteína no llega a la membrana epitelial (clase II) provoca una FQ clásica. La mutaciones que producen CFTR pero que está alterada su regulación (clase III), su conducción (clase IV), o se sintetiza en escasa cantidad (clase V), o están asociadas a una gran variedad de fenotipos con o sin insuficiencia pancreática exocrina.10-11
Hacer el diagnóstico de la enfermedad en la edad adulta podría ser difícil ya sea por la falta de experiencia de los médicos, pues durante muchos años la enfermedad ha sido una entidad eminentemente pediátrica o por la relativa frecuencia de presentaciones leves o atípicas de la enfermedad que pueden pasar inadvertidas o inducir a un diagnóstico erróneo,1como lo fue en nuestro caso en el que no habían antecedentes familiares de la enfermedad, predominaron las manifestaciones respiratorias y no existían manifestaciones digestivas importantes, lo que ha sido reportado como formas larvadas de presentación de la enfermedad.12-13
En este paciente sospechamos la existencia de la FQ por no encontrar la causa que justificara la presencia de Bronquiectasias unido a la infertilidad que presenta el caso y la colonización por Pseudomona Aeuriginosa, variedad mucoide. El diagnóstico clínico se corroboró con el test del sudor con iontoforesis usando pilocarpina.
Aunque nuestro paciente no es portador de 6 de las mutaciones reportadas como las más frecuentes, las que se identifican en 55,5 % de los alelos mutados, pudiera tener otras que afectan también la función de la proteína. No podemos hablar de la correlación entre las mutaciones causales y la expresión clínica de la enfermedad en este caso, pues no sabemos la mutación causal, existen más de1 600 cada una con su expresión fenotípica muy propia. A pesar de que el análisis molecular directo no aportó la información necesaria para el asesoramiento genético, este proceso se llevo a cabo con el resto de los familiares en riesgo, y se les explicó además la probabilidad de recurrencia en esta familia, en la que no se recogen antecedentes de consanguinidad.
En relación con el diagnóstico diferencial, los pacientes con el Síndrome de Young exhiben un cuadro clínico similar a los que presentan FQ, incluyendo bronquiectasias, sinusitis y azoospermia obstructiva. Sin embargo, no presentan incremento de los valores de cloro en el sudor, insuficiencia pancreática, anormal diferencia de potencial nasal, o la mutación delta F508 de la FQ. La artritis reumatoide, y el Síndrome de Sjogren`s (Queratoconjuntvitis sin artritis reumatoidea u otra enfermedad autoinmune concomitante) pueden cursar también con bronquiectasias.
Las bronquiectasias constituyen una rara complicación de otras enfermedades autoinmunes o enfermedades del tejido conectivo, donde se incluye el Lupus eritematoso sistémico, el Síndrome de Marfán, la colitis ulcerativa idiopática y la enfermedad de Crohn (ileítis regional). La Disquinecia Ciliar Primaria (Síndrome de los Cilios Inmóviles), las infecciones pulmonares (Tuberculosis), Micobacterias Atípicas, infección pulmonar por Mycoplasmas, Aspergilosis Alérgica Broncopulmonar, pueden ser otras causas de bronquiectasias, pero no es el caso que presentamos ya que el cuadro clínico y la positividad de los eléctrolitos en el sudor, excluye todas las anteriores.14
En estos momentos, el paciente se encuentra estable llevando tratamiento médico y rehabilitación respiratoria con la Comisión que atiende estos casos en Cuba.
REFERENCIAS BIBLIOGRÁFICAS
1. Ortigosa, L. Cystic fibrosis: Diagnosis. Colomb. Med. [En línea]. 38(1):41-49;2007. (Citado 20 de Enero 2009). Disponible en <http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S1657 5342007000500007&lng=en&nrm=iso>. ISSN 1657-9534.
2. Fanconi G, Vehlinger E, Knauer C. Das Coeliakie-Syndrom bei angeborenem zystischem Pancreas fibromatose und Bronchiek-tasien. Wien Med Wschr . 86: 753-756;1936.
3. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathological study. Am J Dis Chile. 56: 344-349;1938.
4. Tsui LC, Buchwald M, Barker D, Braman JC, Knowlton R, Schrumm JW, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 23: 1054-1057;1985.
5. Rommens J, Iannuzzi M, Kerem B, Drumm M, Melmer G, Dean M, . Identification of the cystic fibrosis gene: Chromosome Walking and Jumping. Science .245: 1059-1065;1989.
6. Riordan J, Rommens J, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science .245: 1066-1073;1989.
7. Kerem B, Rommens J, Buchanam J, Markewicz D, Cox T, Chakrauarti A, et al. Identification of the cystic fibrosis gene: Genetics analysis. Science. 245: 1074-1080;1989.
8. Cystic Fibrosis Genetic Analysis Consortium. (Citado 19 de Enero 2009) URL. Disponible en: http://www.genet.sickkids.on.ca/cftr/app
9. Mata A, Fernando A. Fibrosis quística del adulto. Correlación genotipo-fenotipo pulmonar. Universidad Autónoma de Barcelona. 2006. (Citado 20 de Enero 2009) http://www.tdx.cat/TDX-0412107-152736
10. Girón RM, Arocha BJ. El diagnóstico de la Fibrosis Quística en el adulto.Arch Bronconeumología. 36: 2;2000.
11. Colectivo de autores. Manual de procedimientos de neumología y cirugía torácica. Primera edición.Barcelona:SEPAR;1996, p.1218-1219,v.I.
12. Rindan JR, Rommens JM, Kerem BS, Alon N, et al. Identification of the characterization of complementari DNA. Scienece.245:1066-1072;1989.
13. Ramos CA, Victoria VH, et al. Fibrosis Quística en el adulto. Reporte de un caso. (Citado 19 de enero 2009).Disponible en: www.amc.sld.cu/amc2008/v12n5/amc15508.htm -25k.
14. Katkin JP, MD Clinical manifestations and diagnosis of cystic fibrosis. UptoDate Versión 12.2 2004. CD-ROM.


{kind=link}