










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Habanera de Ciencias Médicas
versión On-line ISSN 1729-519X
Rev haban cienc méd vol.10 no.4 Ciudad de La Habana oct.-dic. 2011
CIENCIAS CLÍNICAS Y PATOLÓGICAS
Instituto Nacional de Oncología y Radiobiología. La Habana. Cuba
Micosis fungoide en estadio tumoral. Presentacion de un caso
Mycosis fungoide in the tumoral stage. Case presentation
Yaniurka Cruz CamejoI; Haslen Hassiul Cáceres LaverniaII; Yoanna Ivette Flores VegaIII; Alicia Amelia Rodríguez AbascalIV; Sofía de la Caridad Alsina SarmientoV; Jorge Luis Morales LópezVI
IEspecialista Primer Grado en Oncología. Calzada San Miguel núm. 8812 entre 1era y Capitán Núñez. Reparto San Juan de los Pinos. San Miguel del Padrón. La Habana. Teléfono: 6916902. Email: burgos@infomed.sld.cu
IIEspecialista Primer Grado en Oncología. Calle 43 núm. 19432 entre 196 y Lindero. La Lisa. Email: haslen.caceres@infomed.sld.cu
IIIEspecialista Primer Grado en Oncología. Línea núm. 1204 entre 18 y 20. El Vedado. Plaza. Teléfono: 8302532. Email: yflores@infomed.sld.cu
IVEspecialista Primer Grado en Oncología. Instructor. Calle 4ta núm. 223 1er piso entre Fomento y Albear. Reparto Palatino. Cerro. La Habana. Email: aliciardguez@infomed.sld.cu
VEspecialista Segundo Grado en Oncología. Auxiliar. Investigador Agregado. Calle 16 núm. 113 entre 13 y 11. El Vedado.
Email: sofialsina@infomed.sld.cu
VIFísico Médico. Línea núm. 1200 entre 16 y 18. El Vedado. Email: gmorales@infomed.sld.cu
RESUMEN
Introducción: La micosis fungoide es una neoplasia maligna originada en los linfocitos T, de curso crónico y caracterizada por lesiones que pueden ser máculas (eritematosas, hipo e hiperpigmentadas), pápulas, placas, parches a veces poiquilodérmicos, nódulos o tumores que pueden ulcerarse y llegan ocasionalmente a la eritrodermia. Tienden a permanecer en la piel por largos períodos de tiempo con lesiones polimorfas aunque en estadios avanzados puede afectar ganglios linfáticos y órganos internos. Aunque sigue siendo la forma más común de todos los linfomas primarios cutáneos de células T (44%), sigue siendo una enfermedad rara. Objetivos: Nos propusimos revisar y analizar la evidencia científica disponible en la literatura mundial para determinar las manifestaciones clínicas más frecuentes de la Micosis fungoide, así como los medios diagnósticos más empleados en la actualidad y su tratamiento. Presentación del caso: Se presentó un paciente de 49 años de edad con hallazgos clínicos e histopatológicos de micosis fungoide en estadio tumoral. Se hace una revisión de la presentación clínica de la enfermedad, su histopatología y tratamiento. Conclusiones: La micosis fungoide sigue siendo una enfermedad rara, sobre todo, en etapa temprana, ya que puede diagnosticarse erróneamente como dermatitis de contacto, psoriasis o dermatitis atópica, por lo que es importante un adecuado diagnóstico.
Palabras clave: Linfoma cutáneo, micosis fungoide.
ABSTRACT
Introduction: Mycosis Fungoides is a malignant neoplasia originated at T- Lymphocytes of chronic course. It is characterized by lesions that could be macules (erythrodermic, hypo or hyper pigmented), papules, plaques, sometimes poikilodermic patches, nodes or tumours that can be festered arriving occasionally to erythroderma. They use to last in the skin for long periods of time with polymorph lesions though in advanced periods they can affect lymphatic ganglions and internal organs. Despite the fact that Mycosis Fungoides is the most common form among the primary cutaneous lymphomas of T cells (44%) it still qualifies as a rare disease. Objectives: We had as principal aim to review and analyze the available scientific evidence in world literature to determine the most frequent clinical manifestations of Mycosis Fungoides as well as the diagnostic means and treatments mostly used at present. Case presentarion: It's a 49 years old patient with clinical and histopathology finds of Mycosis Fungoides in terminal stage. It is made a brief review of the clinical introduction of this disease, its histopathology and treatment. Conclusions: Mycosis Fungoides is still a rare disease mainly in the early stage, that's why it is very important to make an exact diagnosis of it since it can be diagnosed incorrectly as a contact or atrophic dermatitis.
Key words: Cutaneous lymphoma, mycosis fungoides.
INTRODUCCIÓN
Los linfomas cutáneos de células T (LCCT) comprenden una amplia categoría de procesos linfocíticos malignos con afinidad cutánea, preferentemente epidérmica y van a estar caracterizados por una expansión monoclonal de linfocitos T auxiliadores. 1,2 La Micosis fungoide (MF) es la variante más frecuente de estos linfomas (44%), siendo un Linfoma no Hodgkin (LNH) extranodal de células T maduras con compromiso primario cutáneo. 1-4 La placa clásica de MF fue descrita por primera vez, en 1806, por el dermatólogo francés Jean-Louis-Marc Alibert. El nombre de la patología es un tanto engañosa, pues el término vagamente significa «enfermedad fúngica parecida a hongos». 5 Fue llamada así, porque Alibert describió los tumores en piel de un caso grave en un paciente de 56 años de edad como etiología fúngica, y debido a su aspecto lo describió como «un proceso descamativo en la piel y poco después le aparecen tumores por distintas zonas del cuerpo (…) se parecen a hongos, de consistencia como setas». 6 En 1870, Pierre-Antoine-Ernest Bazin describe con mejor detalle la progresión clínica a través de las etapas eczematosa, de placas y de tumores. 7 Posteriormente, Vidal y Broco, en 1885, descubren que pueden aparecer tumores sin que estos sean precedidos por las etapas anteriormente descritas, condición que se denominó con el término Micosis fungoide demblée (D'embleé) o «micosis fungoide primitiva». 8 En 1892, Bernier y Hallopeau reportaron el concepto clínico de Micosis fungoide eritrodérmica, no incluida en la nueva clasificación de la European Organization for Research on Treatment of Cancer (EORTC).3,9 En 1979, un seminario internacional patrocinado por el Instituto Nacional del Cáncer estadounidense acuñó el término LCCT y se emplea en el presente para describir un grupo heterogéneo de linfomas primarios compuesto por células T malignas con manifestaciones en la piel, como la MF. 10 La MF es el subtipo más común de los linfomas de células T. Se estima que su incidencia es 0,29% por cada 100 000 habitantes; es más frecuente en adultos jóvenes, con predominio hombre mujer de 2:1. La regla general de esta enfermedad es que se encuentra limitada a la piel durante muchos años; la diseminación extracutánea sólo puede ocurrir en estadios avanzados y afecta principalmente los nódulos linfáticos, el hígado, el bazo y la sangre. La afectación de la médula ósea es muy rara.11-13 Se ha calculado que su frecuencia de presentación es de más de 80% en mayores de 45 años; sin embargo, entre 4 y 5% ha sido descrito iniciándose en las dos primeras décadas de la vida. 3,4 Su incidencia va en aumento, de acuerdo con los datos reportados por el centro de vigilancia epidemiológica (SEER) siendo de aproximadamente 4 por cada millón, lo que representan 1 500 casos por año.3,14
Es preciso destacar que en la actualidad se describen múltiples variantes atípicas muy diferentes en su presentación clínica a las formas clásicas ya descritas, que establecen la denominación de gran simuladora para esta entidad; es por ello que debemos ampliar nuestro pensamiento clínico y plantear esta posibilidad diagnóstica.1
Se analiza el siguiente caso por ser la micosis fungoide una entidad de presentación no muy frecuente, además por la evolución clínica del caso pasando por las tres etapas en el transcurso del tiempo: etapa de mácula o eritema, etapa de placa y la etapa tumoral; es esta última poco frecuente y la más llamativa en este paciente, tanto por la diseminación de las lesiones como por la respuesta al tratamiento.
DESCRIPCIÓN DEL CASO
Motivo de consulta: Lesiones en piel.
Historia de la enfermedad actual: Paciente masculino, de 49 años de edad. Refiere que hace aproximadamente 20 años comenzó con lesiones máculo-papulosas, pruriginosas de ±1cm, localizadas en ambas regiones axilares; luego, se localizan alrededor de las articulaciones de curso intermitente y son biopsiadas en varias ocasiones con el diagnóstico de psoriasis con seguimiento por Dermatología; recibe tratamiento con esteroides tópicos y orales sin resolución de las lesiones. En diciembre del 2008, adoptan formas de placas, con bordes bien definidos y ,en esta ocasión, se diagnostica como Micosis Fungoide, estadiándose con etapa III A; se planifica quimioterapia (QTP) con esquema CHOP (ciclofosfamida, vincristina, adrimicina y prednisona) 8 ciclos con respuesta parcial culminado en mayo del 2009. Al mes siguiente, reaparecen las lesiones, las que adoptan la forma tumoral, que progresan a pesar del tratamiento con inmunoterapia con interferón (IFN) 3 x 106 IM en días alternos, 28 dosis.
Se realiza revisión de láminas en nuestra Institución y se corrobora este diagnóstico y se informa tumor incompletamente resecado.
Examen Físico (Datos positivos)
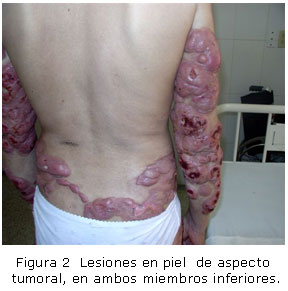
Lesiones en piel de aspecto tumoral, de base ancha, redondeadas, de color rojizo, algunas ulceradas, diseminadas en ambos miembros superiores e inferiores. (Figuras 1 y 2).

Complementarios
Al momento del diagnóstico:
Hto:0.29 Leuco:4 x 109 Plaquetas:200 x 109 V.S.R:95mm/h
T.G.P:39 U.I T.G.O:43 U.I Creatinina: 46.5µmol/l
Estudios imagenológicos
Rx.tórax, U.S abdominal, TAC de tórax y abdomen: Negativos.
Biopsia de Médula ósea: No infiltrada.
Biopsia de piel: Dermatitis de interfase vacuolar donde se observa hiperqueratosis con zonas de espongiosis y queratinocitos necróticos.
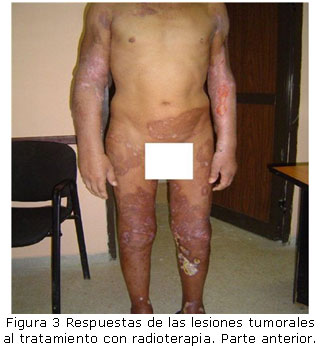
Tratamiento: Recibió tratamiento con radioterapia en el INOR con electrones en LINAC en extremidades inferiores y extremidades superiores. Campo de 40 x 40 cm, dos anteriores y dos posteriores, 3cm de profundidad por variedad clínica tumoral voluminosa. Dosis diaria 200 cGy. Dosis total 30 Gy. La respuesta al tratamiento fue buena. (Figuras 3 y 4).

DISCUSIÓN
Aunque la Micosis fungoide (MF) sigue siendo la forma más común de todos los linfomas primarios cutáneos de células T (44%), es una enfermedad rara. La causa de la MF es desconocida, pero han sugerido el papel de un antígeno crónico (posiblemente un retrovirus), estimulación de linfocitos T helper o CD4 por células de Langerhans intraepidérmica en su génesis. Esta teoría sugiere que la causa de la enfermedad puede ser un proceso proinflamatorio o policlonal. Otra teoría ha sido en relación con el porqué de la progresión de la MF. Esta teoría trata de explicar que la enfermedad se inicia con una fase de exacerbación, en la cual los linfocitos T helper circulantes son reclutados en la dermis papilar, posteriormente ocurre una selección de linfocitos T CD4 por citoquinas, la cual además promueve su proliferación hasta determinadas poblaciones clonales causantes de la neoplasia.11,12,14
Los estadios clínicos se clasifican en estadio de mancha (solitarias o múltiples), estadio de placa (progresión del estadio de mancha, pero puede aparecer de novo) y estadio tumoral, aunque también se han descrito otras variantes clínico- patológicas de la MF, donde puede incluirse la poiquilodermia vascular atrófica, la MF granulomatosa, hipo pigmentada, centro folicular, siringotrópica, d´emblee, de palmas y plantas, la variante ictiosiforme y la enfermedad de Woringer- Kolopp.4,11
Las características clínicas y microscópicas del caso reportado coinciden con el estadio tumoral, que se caracteriza por lesiones tumorales en la piel sana, sobre lesiones no infiltradas o sobre placas infiltradas. Los tumores suelen tener una base ancha, son redondeados o hemisféricos y su tamaño variar. Tienen apariencia de nódulo, son de color rojo brillante y muestran predilección por la cara y los pliegues del cuerpo. En ocasiones, se ulceran y suele ser común que hayan sido precedidos por un estadio de manchas y de placas.
No obstante, pueden manifestarse de nuevo y recibir el nombre de MF d'emblee, que no es lo encontrado en nuestro paciente, pues se describe una larga evolución de la enfermedad.
Desde el punto de vista histológico, en el estadio tumoral se aprecia paraqueratosis y mínimo epidermotropismo. En la dermis papilar y reticular, hay marcado infiltrado de linfocitos atípicos, que pueden mezclarse con células inflamatorias; hay crecimiento vertical de la neoplasia, que puede afectar hasta el tejido celular subcutáneo. Estas células muestran un núcleo involucionado, con patrón de cromatina vesicular.1,4,11
El diagnóstico de linfoma cutáneo exige un abanico amplio de estudios que incluye exámenes clínicos, radiológicos, histológicos y estudios moleculares 14,15 como los realizados a este paciente; se obvian algunos estudios moleculares con los que no contamos en nuestro medio.
De modo general, la característica histopatológica esencial de la MF es la presencia de un infiltrado linfocítico con células medianas y núcleo hipercromático, muy irregular. Estos linfocitos se localizan en la dermis papilar (de forma peri vascular o intersticial) con tendencia a infiltrar la epidermis (exocitosis o epidermotropismo). Es característico que los linfocitos intraepidérmicos se agrupen en microabscesos (de Pautrier-Darier), o bien como células aisladas a lo largo de la unión dermoepidérmica. En cuanto a la dermis, el infiltrado linfocítico se acompaña de fibrosis de la región papilar (signo de los `fetuccini'). Además de linfocitos, las lesiones de MF suelen tener macrófagos y eosinófilos. También están presentes células ínterdigitadas y de Langerhans, células mononucleadas de núcleo hipercromático y forma irregular, conocidas como células atípicas, pudiendo parecerse a las células de Sezary (células de aspecto cerebriforme). De tal modo, que el carácter atípico de estas células, irregularidad en la forma e hipercromatismo nuclear y su epidermotropismo son los dos argumentos o criterios fundamentales para el diagnóstico histológico.4,15,16
El inmunofenotipo de la MF muestra que se trata de un linfoma T periférico constituido por células T (CD3+), helper o cooperadoras (CD4+,CD8-) (ocasionalmente pueden ser CD4-,CD8+)), de memoria (CD45Ro+) que pertenecen al tejido linfoide asociado a la piel (SALT, CLA+) y que además suelen ser CD7- neg. La existencia de células CD30+; sin embargo, pueden indicar transformación maligna.4,15,16
Ocasionalmente, los individuos con MF/SS desarrollan un cambio (transformación) morfológica, condición que puede ser difícil de diferenciar patológicamente de ciertos desórdenes linfoproliferativos, tales como papulosis linfomatoide y linfoma de grandes células anaplásicas Ki-1 positivas.8 Se describen variantes `atípicas' de MF entre estas: variantes hipopigmentada, granulomatosa, reticulosis pagetoide, foliculotrópica, bulosa/vesicular, pustular, púrpura pigmentada like, con mucinocis folicular, acantosis nigricans like, siringotrópica, eritrodérmica, poiquilodermatosa, hiperpigmentada, hiperqueratótica/verrucosa, ictiosiforme, dishidrótica, con transformación a células grandes, dermatitis perioral like, zosteriforme, palmoplantar, vegetante/papilomatosa, pitiriasis liquenoide like y papular. 4,17
Tratamiento
- Las opciones de tratamientos para los pacientes con Micosis fungoide y síndrome de Sézary (MF/SS) incluyen las siguientes:
- Corticoesteroides tópicos.
- Quimioterapia tópica con mecloretamina (mostaza nitrogenada) o carmustina (BCNU).
- Psoraleno y radiación ultravioleta A (PUVA).
- Radiación ultravioleta B (UVB).
- Radiación a toda la piel con haz de electrones (RTPHE).
- Radiación a las lesiones cutáneas sintomáticas.
- Interferón á solo o en combinación con tratamiento tópico.
- Quimioterapia con fármaco único o múltiple.
- Bexaroteno (gel tópico u oral); retinoides.
- Denileucina diftitox (proteína de fusión recombinante de fragmentos de la toxina de la difteria y secuencias de interleucina-2).
- Tratamiento de modalidad combinada.
Estos tipos de tratamientos producen remisión, pero a largo plazo resultan poco comunes. Por tanto, en la mayoría de los pacientes se utiliza tratamiento paliativo, aunque con regularidad se logra una gran mejoría sintomática. De hecho, es común que se logren supervivencias mayores de 8 años. Todos los pacientes de MF/SS resultan idóneos para participar en ensayos clínicos que evalúan nuevos enfoques de tratamiento. Entre las áreas de interés actual en los ensayos clínicos de MF limitados a la piel, figuran terapias de modalidad combinada que contienen fármacos tanto tópicos como sistémicos tales como RTPHE combinada con quimioterapia, mecloretamina tópica o PUVA combinadas con interferón, o técnicas de radiación de campo amplio con PUVA. Hay informes disponibles sobre la actividad de la fotoquimioterapia extracorpórea que usa psoraleno, interferón ã o interferón á; pentostatina, retinoides, fludarabina, aciclovir, 2-clorodeoxiadenosina; seroterapia con anticuerpos monoclonales no marcados, marcados con toxinas o radiomarcados; proteínas de fusión de la toxina ligando del receptor de la superficie celular; y, metotrexato. También se encuentra bajo evaluación, vacunas antígeno- específicas que utilizan células dendríticas y UVB.14,18
Nuestro paciente fue tratado, a lo largo de su enfermedad, antes del diagnóstico definitivo de Micosis fungoide con terapia esteroidea (tópicos y orales), tratamiento que está indicado en esta enfermedad, por lo que quizás obtuvo algún beneficio en la evolución, posterior al diagnóstico de MF se administró quimioterapia con múltiples fármacos, inmunoterapia (interferón alfa) y finalmente radioterapia, generalmente son las terapias disponibles en nuestro medio.
Evolución y pronóstico
El pronóstico de los pacientes con MF/SS se fundamenta en el grado de la enfermedad al momento de presentarse (estadio). La presencia de linfadenopatía y compromiso de la sangre periférica y vísceras suele aumentar al empeorar la afección cutánea y define los grupos de pronóstico precario. La mediana de supervivencia después del diagnóstico varía de acuerdo con el estadio. Los pacientes con enfermedad en estadio I A presentan una mediana de supervivencia de 20 años ó más. En contraste, más de 50% de los pacientes con enfermedad en estadio III y estadio IV mueren de MF; su mediana de supervivencia es menos de 5 años.14,18,19
La evolución natural característica de la MF es lenta. Antes de confirmarse la enfermedad por medio de una biopsia, los síntomas pueden presentarse durante largos períodos, un promedio de 2 a 10 años en forma de erupciones cutáneas crecientes y menguantes, como lo sucedido en el caso presentado que llevaba 20 años con las lesiones en piel, recibiendo tratamiento con esteroides tópicos y orales sin resolución. La MF/SS es tratable con las terapias tópicas o sistémicas disponibles hoy. Sin embargo, hasta ahora ha resultado difícil encontrar modalidades curativas, con la posible excepción de aquellas para pacientes con enfermedad mínima limitada a la piel. 14,18,19
La enfermedad cutánea por lo general evoluciona de un estadio de parche o placa eccematosa que cubre menos de 10% de la superficie corporal (T1), a un estadio de placa que afecta 10% o más de la superficie corporal (T2) y, finalmente, evoluciona a tumores (T3), que con frecuencia experimentan ulceración necrótica. El SS es una forma avanzada de MF con eritrodermia generalizada (T4) y compromiso de la sangre periférica al momento de presentarse. Una causa común de defunción durante la fase tumoral es la sepsis causada por Pseudomonas aeruginosa o Estafilococcus aureus, debido a infecciones crónicas de la piel con especies estafilocóccicas e infecciones sistémicas posteriores.14,18,19
CONCLUSIONES
Nos ha parecido de interés comunicar este caso clínico, ya que demuestra la importancia del diagnóstico diferencial de Micosis fungoide, especialmente en etapa temprana, ya que puede diagnosticarse erróneamente como dermatitis de contacto, psoriasis o dermatitis atópica, por lo que el estudio histológico es sumamente importante y si este no es concluyente deberán realizarse biopsias cada tres meses.
REFERENCIAS BIBLIOGRÁFICAS
1. Collazo Caballero SE. Micosis fungoide y el síndrome de sézary. Diagnóstico, Estadiaje y Tratamiento. [Manual de prácticas médicas]. HCQ H Ameijeiras: 2da ed. La Habana; 2008.
2. Beltrán Gárate B, Sánchez Félix G, Morales Luna D, Castro Vargas G, Phillco Salas M, Paredes Arcos A, et al. Consenso Peruano de Diagnóstico y Tratamiento de la Micosis Fungoides y Síndrome de Sezary. Acta Med Per. 2009; 26 (3):180-3.
3. Rodríguez Acar M, Guzmán Vázquez OE. Micosis fungoide: comunicación de un caso. Rev Cent Dermatol Pascua. May-Ago 2004; 13 (2):95-8.
4. Vidarte Orrego G, Álvarez Llanos EG. Micosis fungoide en estadio de placa. Dermatol. Perú. 2008; 18(2):118-121.
5. Darrell R, Friedman R, Dzubow LM, Reintgen DS, Bystryn JC, Marks R. Cáncer ofthe Skin. 1st ed. España: Elsevier; 2006.
6. Garzona Navas L, Moreira Hidalgo F, Hidalgo Matlock B. Micosis Fungoide: Revisión de tema y presentación de un caso. Rev Costarric salud pública. 2007; 16 (30): 46-53.
7. Du Vivier An, McKee PH. Atlas of clinical dermatology. 3rd ed. Elsevier Health Sciences; 2002.
8. Van Doorn R, Van Haselen ChW, Van Voorst PC, Geerts ML, Heule F, De Rie M, et al. Mycosis fungoides. Disease evolution and prognosis of 309 dutch patients. Arch Dermatol. 2000; (136):504-10.
9. Reyes Flores O. Los «disfraces» de la micosis fungoide. Dermatología Venezolana. 1996; 34 (1):3-4.
10. Pinter-Brown, Lauren C. Mycosis fungoides. Stem Cells and Disorders ). Oct 2008; eMedicine.com. [Cited 2009 Jun 28] .Available from: http://www.ncbi.nlm.nih.gov/books/Mycosis fungoides
11. Juárez-Navarrete L, Rincón-Pérez C. Linfomas cutáneos: Fisiopatología y Clasificación. Dermatol Rev Mex. 2005; (49):109-22.
12. Odom R, James W, Berger T. Andrew's Dermatologia Clínica. España: Editorial Marban Libros SL; 2004.
13. Torres V, Camacho F, Mihm M, Sober A, Sánchez Carpintero I. Dermatología Práctica Ibero latinoamericana. 1era ed. Cali:Vicente Torres Lozada Nieto; 2005.
14. DeVita VT, Lawrence ThS, Rosenberg SA. Devita, Hellman and Rosenbergs Cancer: Principles and Practice of Oncology. 8th ed. Lippincont Williams and Wilkins; 2008.
15. Panta Hidalgo S. Micosis fungoide. Rev Soc Ecuat Dermat [Internet]. 2002 Aug. Available from: http://www.dermatologiaecuatoriana.com
16. Jaffe ES, Harris NL, Stein H, Isaacson PG. Classification of lymphoid neoplasms: the microscope as a tool for disease discovery. Blood [Internet] . 2008; (112):4384. Available from: http://www.uv.es/derma
17. Vicuña C, Paredes A, Carvajal T, Revollar Y, Sánchez G. Variantes atípicas de micosis fungoide: folículo trópica e hiperpigmentada. Dermatol Per. 2005; 15(3).
18. Instituto Nacional del Cáncer [Internet]. Información general sobre la micosis fungoide y el síndrome de Sézary: Tratamiento (PDQ) (en español). [Actualizado 2010 Nov 24; Citado 2011 Feb 14]. Disponible en: http://www.cancer.gov/español
19. Aguilar García CR, Morales Guadarrama R, Luna Álvarez H, Jiménez Jiménez E. Micosis fungoide. Presentación de un caso y revisión de la bibliografía. Med Intern Mex. 2009; 25 (4): 317-20.
Recibido: 15 de junio de 2011.
Aprobado: 1 de noviembre de 2011.

