










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La anoftalmia congénita es una malformación ocular caracterizada por la ausencia total de tejido ocular dentro de la órbita. Esta entidad fue reportada por primera vez por Lyscostenes y Scenck en 1609 y a inicios del siglo XIX Briggs descubrió el carácter hereditario de esta anomalía al diagnosticarla en siete casos de una misma familia.1
La anoftalmia es la malformación ocular congénita más grave. Su incidencia se ha determinado en 1 por cada 30,000 nacimientos2 y la prevalencia de esta entidad al nacimiento se ha estimado en 3 por cada 100.000 habitantes y hasta 30 por cada 100 000 habitantes cuando está asociada a otras malformaciones3 y representa aproximadamente 4% de las causas de ceguera congénita junto a la microftalmia, con la cual se asocia en las descripciones bibliográficas y en la mayoría de los síndromes malformativos, por tal motivo, es común que se presenten de manera conjunta estas dos malformaciones oculares describiéndolas como microftalmia/anoftalmia.4
La microftalmia congénita por su parte, es la reducción variable del volumen del globo ocular, Su frecuencia es mayor que el anoftalmo. Estados Unidos reporta entre 0,18 - 0,4 por cada 10 000 nacidos vivos respectivamente.1 Esta afección puede ser simple o compleja. La simple se refiere a un ojo completo pero pequeño y en la compleja se asocia a diversas malformaciones de varias partes del ojo, además de su tamaño pequeño.5
Se conoce que el examen ocular en los primeros días de vida puede presentar dificultades para realizarlo por el edema de los párpados propio de las primeras horas y porque los recién nacidos tratan de mantener los ojos cerrados ante los intentos de abrirlos o por la exposición a la luz,6 pero el reconocimiento adecuado del sistema visual, en el periodo neonatal antes del egreso de la maternidad, reviste gran importancia pues el diagnóstico temprano de las alteraciones oculares permite tomar una conducta anticipada que puede ser quirúrgica o no y permitir también el diagnóstico de síndromes genéticos. Para el diagnóstico de esta malformación se requiere realizar una evaluación interdisciplinaria donde participen oftalmólogos, genetistas neonatólogos e imagenólogos, que orientarán los estudios, la evaluación y el tratamiento,7,8 entre estos estudios se encuentra la ultrasonografía ocular, la Tomografía Axial Computarizada (TAC) y la Resonancia Magnética Nuclear (RMN), esta última, es la técnica de elección en el diagnóstico preciso de las malformaciones congénitas de cualquiera de las estructuras del sistema nervioso central,9 dado su elevado grado de especificidad e inocuidad para este grupo de edad.
En la actualidad las imágenes médicas se han convertido en un instrumento fundamental de la práctica clínica ya que permiten detectar enfermedades con una gran precocidad y precisión.10
Objetivo
Teniendo en cuenta todo lo antes expuesto se decidió realizar la presentación de este caso con el objetivo de demostrar la importancia de la neuroimagen en el diagnóstico y orientación de la microftalmia/anoftalmia neonatal congénita bilateral.
Presentación del caso
Recién nacido femenino hijo de madre de 24 años de edad con serología no reactiva y grupo sanguíneo A positivo con antecedentes de sepsis vaginal durante el embarazo sin otros antecedentes que nace a las 40,3 semanas de edad gestacional por parto eutócico con líquido amniótico claro, con rotura prematura de membranas ovulares de 32 horas, presentación cefálica, evaluación de Apgar 9/9 y peso al nacer de 3 000 gramos.
Durante el examen físico inicial realizado en la primera hora de vida por especialista de neonatología, se constata como datos positivos hipertelorismo, orejas de implantación baja, fisura palatina, ano anterior y ausencia de los globos oculares. No se detectaron alteraciones genitales.
Antes de las primeras 24 horas de vida se interconsultó con especialista de oftalmología quien al examen clínico reportó hipertelorismo, ligera fimosis palpebral, hendidura palpebral pequeña, entropión y ausencia de los globos oculares a la palpación y al examen físico instrumentado.
A las 36 horas de vida se efectuó ultrasonido ocular confirmándose el diagnóstico de anoftalmia/microftalmia al visualizar a través de esta técnica, estructuras intraorbitarias ecolúcidas, ovoideas, pequeñas bien precisadas y rudimentarias, así como imágenes de estructuras ecogénicas semicirculares de centro anecoico bilaterales excéntricas, compatibles con invaginación inconclusa del ectodermo que da origen a una falla en la formación de la vesícula óptica. No pudiendo definir las imágenes habituales de globos oculares por lo que se sugirió por radiólogo realizar RMN.
En ultrasonido transfontanelar se mostró hipoplasia del cuerpo calloso, cuernos posteriores dilatados y aumento del espacio subaracnoideo en fosa posterior.
No se observaron alteraciones anatómicas en ultrasonido abdominal.
A las 48 horas de edad el ecocardiograma no mostró anomalías estructurales.
Se le tomaron muestras para estudios del complejo TORCHS (Toxoplasma, Rubeola, Citomegalovirus, Herpes Virus, Sífilis) y PCR TR (reacción de cadena de polimerasa en tiempo real, por sus siglas en ingles) en los estudios del virus Zika, que fueron negativos.
Al 4to día fue evaluado por especialista en maxilofacial que corroboró el diagnóstico de fisura palatina completa.
Fue evaluado por genetista clínico al 5to día de edad no concluyendo un diagnóstico definitivo y orientó realizarle estudios cromosómicos posteriormente.
A los 8 días de vida la RMN mostró los siguientes resultados: Esbozos de lentes vesiculares rudimentarios ubicados en zonas atípicas por conflicto de espacio dentro de las órbitas con múltiples imágenes Hipointensas en T1 e Hiperintensas en T2 de aspecto quístico, adyacentes, de interface de pared definida, libres de elementos en suspensión, bilaterales y esbozo de nervio óptico del lado derecho dada su prolongación lineal, longitudinal, más allá del límite posterior de la órbita derecha en planos de cortes axial y sagital respectivamente, sin definirse imágenes de globos oculares. (Figuras 1, 2 y 3).
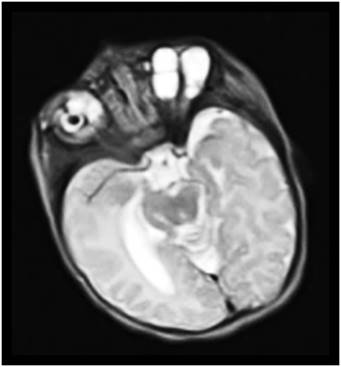
Fig. 1 Corte axial Resonancia Magnética Nuclear T2. Estudio realizado en Hospital Pediátrico Docente “Juan Manuel Márquez”. La Habana, Cuba
Esbozos de lente vesicular rudimentario ubicados en zonas atípicas por conflicto de espacio dentro de la órbita Izquierda. Múltiples imágenes Hiperintensas en T2 de aspecto quístico, adyacentes, de interface de pared definida, libres de elementos en suspensión órbita derecha.
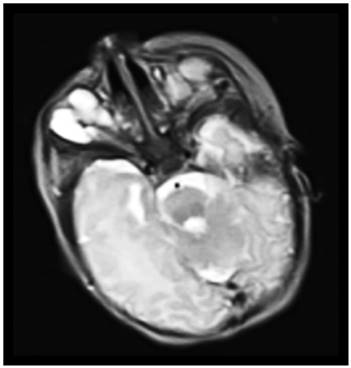
Fig. 2 Corte axial Resonancia Magnética Nuclear T2. Recién nacido. Estudio realizado en Hospital Pediátrico Docente “Juan Manuel Márquez”. La Habana, Cuba
Múltiples imágenes Hiperintensas en T2 de aspecto quístico, adyacentes, de interface de pared definida, libres de elementos en suspensión.

Fig 3 Corte sagital Resonancia Magnética Nuclear T2. Recién nacido. Estudio realizado en Hospital Pediátrico Docente “Juan Manuel Márquez”. La Habana, Cuba
Esbozos de lente vesicular rudimentaria ubicado en zonas atípica por conflicto de espacio dentro de la órbita derecha con múltiples imágenes Hiperintensas en T2 de aspecto quístico, y esbozo de nervio óptico dada su prolongación lineal, longitudinal, más allá del límite posterior de la órbita derecha en plano de corte sagital, sin definirse imágenes de globos oculares.
Durante la estadía hospitalaria no presentó convulsiones, hipotonía, ni otras alteraciones neurológicas.
A los 18 días fue egresado de la maternidad con lactancia materna exclusiva, con seguimiento en consulta por especialista en maxilofacial, remisión a consulta de baja visión para implante de prótesis intraorbitarias y seguimiento por consulta de neurodesarrollo del centro desde donde se dirigirán las interconsultas con las diferentes especialidades implicadas en este caso.
Discusión
Este paciente es portador de una malformación congénita, con patrón de defectos congénitos múltiples en el cual era muy difícil el diagnóstico de una alteración ocular aislada, por lo que fue necesario realizar estudios imagenológicos que determinaran la presencia de otras alteraciones como refiere Bermejo Sánchez,4 en este caso, la ultrasonografía, que es un proceder diagnóstico inocuo, barato y muy accesible, el cual incluso puede repetirse en múltiples ocasiones9 y la indicación de otros medios diagnósticos de neuroimágenes como la resonancia magnética nuclear que resulta ser uno de los ideales y más sensibles en la demostración posnatal de las anomalías por medio de imágenes multiplanares que ofrece resultados en diferentes secuencias de cortes e intensidades, lo que permitió la descripción con exactitud y especificidad la afectación ocular, así como de la existencia de otras lesiones congénitas asociadas.11
En este paciente se descartó que esta entidad fuera secundaria a una embriofetopatia por no recogerse el antecedente materno de diabetes ni el consumo de alcohol, infecciones no específicas, deficiencia de Vitamina A o uso de medicamentos teratogénicos como refieren otros estudios.4 Dentro de las embriofetopatias también se descartaron las infecciones prenatales al realizar los estudios correspondientes para Toxoplasma Gondii, la cual puede ser causa de Anoftalmia / Microftalmia y otras malformaciones congénitas como describió Arce-Estrada.12) También se descartó el virus del Zika que se ha mencionado como causa de malformaciones oculares, como parte de un síndrome congénito.13
Entre los síndromes cromosómicos relacionados con esta malformación congénita ocular se encuentran la Trisomía 13, la 18, la 21 y Monosomía X, Aneuploidías del Y además de otras alteraciones estructurales, por lo que es importante en este paciente realizar estudio del cariotipo posteriormente9 como orientó la especialista en genética, aunque este paciente no presentó características fenotípicas evidentes que orientaran el diagnóstico de las alteraciones cromosómicas 13 (Síndrome de Patau), 18 (Síndrome de Edwards) y 21 (Síndrome de Down), se deberá esperar el estudio cromosómico para descartarlas totalmente.14
En este caso se constató que la malformación es de tipo compleja porque se presentó asociada a otros defectos congénitos, lo cual puede verse tanto en síndromes como en otros tipos de cuadros polimalformativos genéticos.4 La Anoftalmia/Microftalmia puede estar asociada a múltiples síndromes genéticos no cromosómicos, entre ellos pudiera corresponderse con el Síndrome De Morsier, teniendo en cuenta los hallazgos en la RMN con manifestaciones de Displasia Septo-óptica que en este caso, como el presentado por Aviña,15 no se demostró afectación endocrina del eje hipotálamo hipofisario en el periodo neonatal durante el tiempo que estuvo hospitalizado, además que no se mostró alteraciones estructurales de la hipófisis en la RMN la cual es de gran valor en el diagnóstico de las alteraciones de la forma y el tamaño de esta estructura.16
Desde el punto de vista clínico se realizó diagnóstico diferencial con el síndrome de Lenz pues este presenta colobomas, cardiopatías, atresia de coanas, anomalías de los genitales y malformaciones de las manos como polidactilia, clinodactilia o sindactilia, también se descartó el Síndrome de Fryns que se acompaña de hernia diafragmática y alteraciones de las falanges, el síndrome de Matthew-Wood que se acompaña de hipoplasia pulmonar, el síndrome de Walter-Warburg que se acompaña de distrofia muscular y alteraciones cerebelosas y el síndrome oftalmo-acromélico de Waardenburg que tiene deformidades en extremidades y retardo mental severo, aspectos clínicos que no presentó el paciente evaluado.15,16,17
Los autores de este trabajo recomiendan realizar seguimiento en consulta multidisciplinaria de neurodesarrollo a los pacientes con Anoftalmia / Microftalmia donde se evalúe su desarrollo psicomotor y las posibles afectaciones de otras esferas sensoriales que pueden estar afectadas en los diferentes síndromes que pueden acompañar esta malformación ocular, además se deberá evaluar con especialista en endocrinología para realizar estudios que descarten las diversas manifestaciones de acuerdo con las hormonas del eje hipotálamo-hipofisario que pueden hallarse deficitarias como la hormona del crecimiento, la antidiurética, la adenocorticotropa, la liberadora de tirotropina, la liberadora de gonadotropina y la liberadora de hormona del crecimiento, entre otras16 que requieran alguna intervención terapéutica y además recomiendan realizar RMN a todos los recién nacidos con este tipo de malformaciones oculares.

