










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El Síndrome de Sanfilippo o MPS III es un error innato del metabolismo reconocido por el pediatra norteamericano Sylvester Sanfilippo en 1963.1 Presenta un patrón de herencia autosómico recesivo y es causado por la deficiencia de una de las cuatro enzimas involucradas en el catabolismo del heparán sulfato (HS); en función de esto, existen cuatro subtipos de esta entidad (MPS III A, B, C, D). Este Glicosaminoglicano (GAG) se acumula progresivamente en diferentes órganos y tejidos lo que explica en gran medida su carácter multisistémico, aunque predomina la afectación del sistema nervioso.2,3,4
La MPS III B tiene como base molecular el déficit de la enzima N-acetil αD- glucosaminidasa, cuyo gen NAGLU se encuentra ubicado en el locus 17q21.2.5) Se caracteriza clínicamente por la presencia de facie ligeramente tosca, sinofris, infecciones respiratorias frecuentes, deterioro neurológico de curso crónico y progresivo, trastorno de conducta y pérdida de habilidades adquiridas.2,6
La incidencia para MPS III se estima entre 1:50 000-1:250 000 nacidos vivos; existe una gran variación entre los datos publicados para distintas regiones del mundo.7
En 1996, se determinó la estructura del gen NAGLU por Zhao y cols., desde entonces, múltiples han sido las variantes alélicas reportadas en las bases de datos internacionales para este gen.8,9) En Cuba, desde 1985 se han diagnosticado enzimáticamente siete pacientes.10) Solo en tres casos ha sido posible identificar las variantes alélicas responsables de la enfermedad, uno de los cuales es el paciente que se describe.
Se presenta este caso, con el objetivo de describir las manifestaciones clínicas, bioquímicas y moleculares de un paciente con Síndrome Sanfilippo B, en quien se confirma una mutación no descrita anteriormente.
Presentación del caso
Para el diagnóstico del paciente se aplicó el método clínico, a través de la técnica comparativa o de patrón y se confirmó a través de estudios bioquímicos y moleculares.
Aspectos Éticos: Se obtuvo la aceptación por parte de los padres para el estudio, publicación de los resultados y fotografía del paciente.
Caso clínico. (Figura 1).
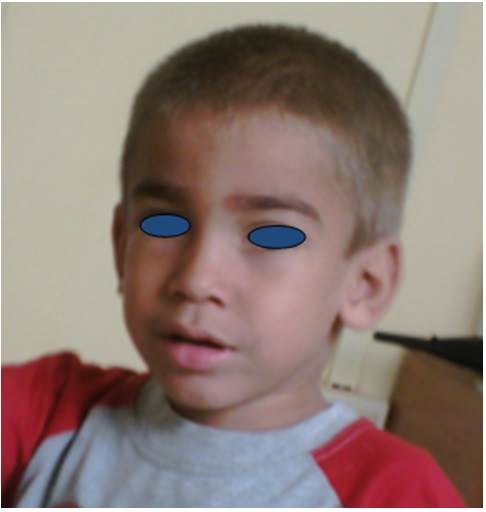
Fig.1 Paciente con Síndrome Sanfilippo B a los 3 años de edad. Nótese la facie ligeramente tosca, cabello y cejas gruesas
Paciente de 13 años, masculino, hijo de padre y madre jóvenes sanos (31 y 29 años, respectivamente) con una hermana de 18 años aparentemente sana; acude a consulta de genética clínica de la ciudad de Camagüey, remitido por el pediatra del área de salud, por presentar infecciones respiratorias a repetición y hepatomegalia. En la familia, no es referida la existencia de consanguinidad ni la presencia de antecedentes patológicos de interés.
Antecedentes prenatales: Embarazo 2, Parto 2, Aborto 0, estudios ultrasonográficos normales, valor normal de alfafetoproteína.
Antecedentes perinatales: Parto normal, tiempo de gestación 38.5 semanas, Apgar 9/9, peso 3 280 g, talla 50 cm.
Antecedentes postnatales: neumonía congénita.
Signos Clínicos presentes en el paciente. (Tabla 1)
Tabla 1 Signos clínicos descritos en el Síndrome Sanfilippo B. Presencia o ausencia en el paciente
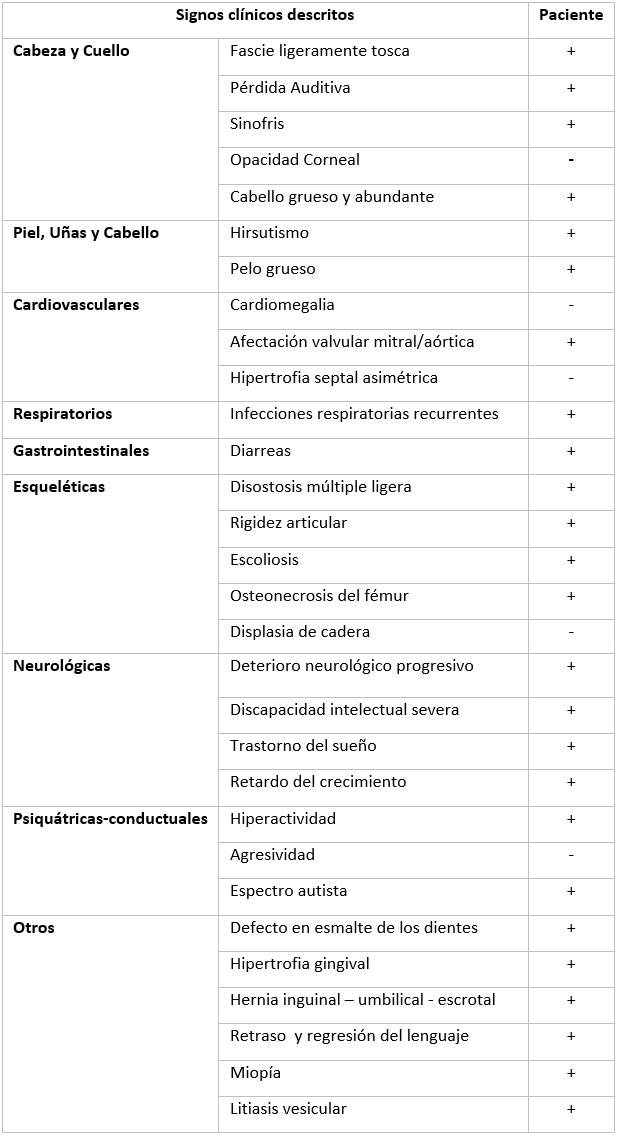
Leyenda: (+) Signo presente; (-), signo ausente.
Alimentación: Lactancia materna exclusiva hasta los cuatro meses, aún así presentaba deposiciones diarreicas sin explicación.
En su primer año de vida presentó procesos respiratorios inflamatorios en varias ocasiones, requiriendo tratamiento repetido con antimicrobianos, se mantenían los cuadros diarreicos cíclicos sin causa aparente.
En la etapa transicional continuó con cuadros respiratorios infecciosos que se hicieron más frecuentes, asociados a otitis media y adenoiditis crónica, siendo la respiración ruidosa. Lograba el sueño tarde e intranquilo, se evidenció ligero retardo en el desarrollo psicomotor, pero no en el lenguaje; además no tenía control de esfínteres y se describió a esta edad litiasis vesicular al realizar examen ultrasonográfico. El paciente logró caminar a los 15 meses de edad.
En la etapa preescolar la conducta fue hiperactiva, aparecieron rasgos de autismo; fue evaluado por logopedia por presentar trastorno en el lenguaje, donde se concluyó la presencia de dislalia funcional. Paralelamente, mediante ecografía abdominal se visualizó la presencia de hepatoesplenomegalia. Posteriormente presentó un sangramiento digestivo bajo debido a colitis ulcerativa y pólipos intestinales, por lo cual fue manipulado bajo tratamiento anestésico. Después de la conducta anterior, se detectó una regresión total del lenguaje.
En la etapa escolar presentó mayor deterioro neurológico y de conducta, trastorno en el sueño eventual, afectación cardiovascular con insuficiencia valvular mitral ligera y Enfermedad de Perthes, lo cual dificultó su deambulación. (Figura 2).
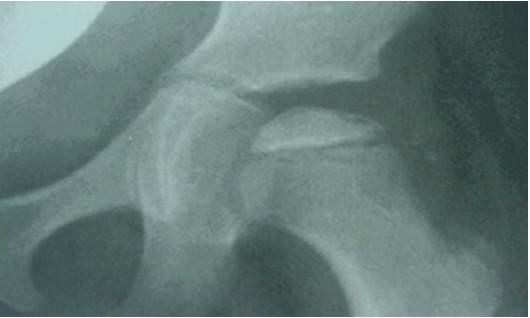
Fig. 2 Radiografía de pelvis donde se muestran signos radiográficos de Enfermedad de Perthes. Obsérvese aumento de la densidad de la epífisis, pérdida del espacio articular, línea radiolucente a nivel de la cabeza femoral por fractura subcondral secundario al colapso óseo
Las infecciones respiratorias, frecuentes, requirieron tratamiento con antimicrobianos.
Actualmente presenta mayor deterioro neurológico, mantiene conducta destructiva pero no agresiva, trastorno del sueño eventual, deambula con mucha dificultad, requiere de la ayuda y cuidado de otra persona, las infecciones respiratorias son más frecuentes.
Se realizaron estudios radiográficos del cráneo, tórax, columna y pelvis, ecografía de abdomen y área cardiovascular. Como resultados de estos estudios se confirmó: cifoescoliosis dorsal, signos radiográficos de Enfermedad de Perthes, hepatomegalia de 3 cm, litiasis vesicular, múltiples, ligera esplenomegalia. Insuficiencia mitral moderada.
Para el diagnóstico bioquímico, se emplearon los métodos colorimétricos test de Berry, la reacción de precipitación con el bromuro-cetil-trimetil-amonio (BCTA) y la cromatografía en placa delgada, para identificar la presencia de GAG urinarios. Posteriormente se obtuvo 10 ml de sangre venosa mezclada con 150 µl de heparina sódica del paciente, los padres y un control aparentemente sano sin antecedentes patológicos para enfermedades de depósito lisosomal ni vínculos familiares. A partir de estas muestras se obtuvieron homogenados de leucocitos para la cuantificación de la actividad relativa de la N-alfa-acetilglucosaminidasa. Para ello fueron empleados los procedimientos descritos internacionalmente.11) Para el estudio molecular se utilizó ADN genómico del paciente y sus padres obtenido a partir de sangre periférica mediante el método de precipitación salina.12) La identificación de las mutaciones se realizó empleando secuenciación masiva utilizando el panel de secuenciación de exoma clínico comercializado por Illumina (TruSightOne, por sus siglas en inglés) y la captura virtual de los genes para su análisis bioinformático. Las variantes seleccionadas se confirmaron mediante la secuenciación convencional de Sanger empleando secuenciador Terminator de BigDye (AppliedBiosystems, Foster City, CA) usando ADN genómico de los pacientes y el de sus padres.13,14) Se realizó predicción funcional de la variante alélica “in silico” empleando MutationTaster.
Los resultados de estudios bioquímicos y moleculares se describen en la Tabla 2.
Tabla 2 Resultados de estudios bioquímicos y moleculares realizados
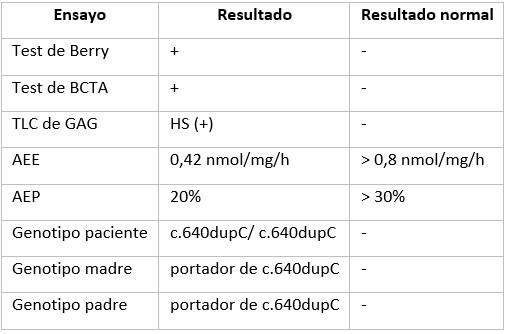
Leyenda: (+): positivo; (-): negativo; AEE: actividad enzimática específica de N-acetil αD- glucosaminidasa.; AEP: actividad enzimática porcentual
Este paciente es evaluado periódicamente por Neurología, Otorrinolaringología, Gastroenterología, Cardiología, Oftalmología, Fisiatría, Logopedia, Ortopedia y Genética Clínica.
Discusión
El Síndrome Sanfilippo B es uno de los cuatro subtipos de MPS III que se caracteriza por degeneración progresiva del sistema nervioso central, regresión del desarrollo, junto a otras manifestaciones neurológicas: espectro autista, problemas de conducta y alteraciones del sueño, lo cual contrasta con la relativa discreta repercusión somática, en el orden esquelético, visceral y facial.15,16,17
El depósito gradual y progresivo de HS, característico en personas con este defecto, conduce a una amplia variabilidad en la expresión clínica, que incluye fenotipos severos - con gran afectación de la calidad de vida - y otros ligeros, que en ocasiones pueden escapar o retardar el diagnóstico.18
Esta gran variación descrita entre los diferentes subtipos de MPS III está dada por la gran heterogeneidad alélica y no alélica que presenta esta enfermedad.2,18
A pesar de la diferencia en la alteración bioquímica presente, los cuatro subtipos clínicamente indistinguibles, tienen como signo principal el deterioro neurológico progresivo; por tal motivo se les consideran como paradigma de las manifestaciones de acúmulo de GAG en el sistema nervioso, característica que lo distingue del resto de las MPS.16,18
Las manifestaciones somáticas, comunes a otras MPS, aunque ligeras en la MPS III, también pueden verse y formar parte de su amplio espectro clínico - con repercusión cardiovascular, gastrointestinal, esquelético, respiratorio, ocular y auditivo.14,19
La evolución y aparición de las manifestaciones clínicas son referidas, por varios autores, en tres estadios:
Estadio I: Generalmente en etapa transicional, previo al diagnóstico, presentan retardo en el desarrollo psicomotor, afecta primariamente el lenguaje.
Estadio II: En esta segunda etapa (3-10 años) predominan las alteraciones en el comportamiento: hiperactivo, ansioso, agresivo y destrucçtivo. La alteración en el sueño: insomnio, sonambulismo, inversión del ciclo día- noche está presente hasta en 90 % de los casos. Se hace más evidente la afectación en el aprendizaje, lenguaje y comprensión.
Estadio III: Después de la primera década, se observa una regresión del intelecto, lenguaje y área motora. Hay pérdida continua de habilidades y lento deterioro hasta llegar a un estado vegetativo con dependencia total y muerte finalmente.
Sin embargo no todos los pacientes tendrán el mismo patrón de deterioro.5,7
Otros autores prefieren clasificar esta enfermedad de acuerdo con su severidad en tres grupos principales: fenotipo severo: aquellos que en la adolescencia dependen totalmente del cuidado de personas; fenotipo intermedio: aquellos que tienen una regresión de habilidades más lento y viven en la adultez; fenotipo atenuado: capaces de alcanzar un nivel de desarrollo más alto y lograr mantener el lenguaje y la deambulación en la adultez.2,14
El diagnóstico de MPS III se sospecha teniendo en cuenta las manifestaciones clínicas y la mucopolisacariduria exclusiva de HS. El diagnóstico diferencial debe establecerse con otros desórdenes de acúmulo lisosomal, de ellas las MPS I, II y Mucolipidosis II.6) La disminución de la actividad enzimática específica y/o la identificación de variantes alélicas patogénicas en NAGLU, son criterios suficientes para su confirmación.1,2
Las características presentes en el paciente coinciden con lo descrito en la literatura, donde predomina la progresiva degeneración neurológica junto a las alteraciones de conducta que ha ido atravesando por distintas etapas: hiperactiva, autista, destructiva. También, secundarias al compromiso del sistema autónomo, hay presencia de diarreas; lo que está descrito en la tercera parte de los pacientes. Las infecciones respiratorias altas y bajas, constituyen evidencia precoz de MPS III. Sin embargo, el trastorno del sueño, signo frecuentemente descrito en estos pacientes, solo se presentó en las primeras etapas de la vida, siendo actualmente ocasional. La valvulopatía mitral y la Enfermedad de Perthes, menos descritas, también forman parte de la evolución de la enfermedad que afecta su calidad de vida.5,7,15,16,17
El sangramiento digestivo provocado por la colitis ulcerativa y pólipos intestinales no ha sido descrito en otros casos, según la literatura revisada, hallazgo que puede estar relacionado con su condición, dado lo infrecuente de estas patologías en la niñez.19,20
Los resultados obtenidos en los ensayos colorimétricos y cromatográficos junto a su evaluación clínica, contribuyeron a elevar la sospecha diagnóstica de MPS III. Posteriormente, la disminución demostrada en la actividad enzimática de la N-Acetil αD-glucosaminidasa confirmó el diagnóstico bioquímico de MPS IIIB.
Finalmente, la secuenciación del ADN, permitió confirmar la presencia de la variante alélica en el gen NAGLU c.640dupC; p.L214Pfs*59 en homocigosis (NM_000263.3; NP_000254.2). Al identificar que cada uno de los padres son portadores de esta mutación, se pudo demostrar la segregación Mendeliana de estos alelos y que la condición de homocigosis del paciente no es debido a una disomía uniparental. Esta variante no ha sido encontrada en las bases de datos de mutaciones puntuales más importantes: ExAC, en 1 000 Genome Project, en Clinvar, HGMD o LOVD3; por lo que se considera una nueva mutación para NAGLU.9,20,21,22
El estudio “in silico” mostró reales probabilidades de causar la enfermedad (mutación patogénica) por afectar severamente la estructura de la proteína; esto es debido a una alteración en la secuencia de aminoácidos por corrimiento del marco de lectura, y provoca la aparición temprana de un codón de terminación que conlleva a la síntesis de una cadena de 272 aminoácidos en lugar de 743, además de afectar sitios de eliminación de intrones y varios sitios de N-glicosilación de dicha proteína.23
En el momento actual de la enfermedad del paciente, las infecciones respiratorias son las principales causas de morbilidad, lo cual afecta y deteriora progresivamente su calidad de vida; requiere cuidado permanente de otra persona pues deambula con mucha dificultad secundario a la regresión propia de la evolución de esta severa afección. Con todo esto, a pesar del excelente medio familiar que rodea al paciente, lo cual contribuye positivamente en su calidad de vida, el curso de esta enfermedad definitiva e invariablemente es progresivo y fatal.
En efecto, como puede observarse, la mutación identificada presumiblemente severa, coincide con la expresión fenotípica también severa de esta enfermedad.
Similares resultados obtuvieron Weber y cols, en estudios de correlación genotipo-fenotipo realizado a pacientes con MPS IIIB, a quienes identificaron múltiples mutaciones que provocaban terminación prematura o elongación del producto génico, todas asociadas a fenotipo severo.4,9,19
Finalmente se debe resaltar la gran repercusión preventiva que tiene en el asesoramiento genético de la familia, confirmar el diagnóstico de esta enfermedad a través del estudio molecular. Este resultado contribuye a realizar la correlación genotipo-fenotipo y ofrecer un pronóstico; permite además sospechar la posibilidad de consanguinidad entre ambos padres -dada la homocigosidad de la mutación y procedencia familiar de igual región-, así como realizar el diagnóstico prenatal, preimplantación y de portador en otros miembros de la familia.
Conclusiones
En este paciente, predominaron las alteraciones de conducta, deterioro neurológico progresivo e infecciones respiratorias, Se identificó la presencia de colitis ulcerativa y pólipos intestinales, los que fueron hallazgos no descritos anteriormente para esta enfermedad. Esta se confirmó desde el punto de vista enzimático y molecular; se detectó una variante alélica patogénica no descrita en ningún otro paciente con MPS IIIB del mundo. Se resalta la repercusión preventiva del diagnóstico molecular en el asesoramiento genético a la familia.

