










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los síndromes de poliposis hamartomatosos constituyen entidades raras que se caracterizan por el desarrollo de pólipos hamartomatosos en el tracto gastrointestinal los cuales incluyen, fundamentalmente los síndromes de Peutz Jeghers (SPJ), poliposis juvenil, tumor hamartoma, PTEN (síndrome de Cowden, síndrome de Bannayan-Riley-Ruvalcaba y Proteus), y poliposis mixta hereditaria.1
El SPJ se caracteriza por la presencia de pólipos hamartomatosos en el tracto gastrointestinal, hiperpigmentación mucocutánea y predisposición a presentar cáncer.2 Tales pólipos pueden presentarse también en sitios extraintestinales como pelvis renal, bronquios, vesícula biliar, conductos nasales, vejiga urinaria y uréteres.3 Las máculas hipermelánicas aunque no patognomónicas se presentan en regiones periorificiales, y en palmas y plantas de manos y pies.4,5) El dolor abdominal, sangrado gastrointestinal, anemia secundaria, prolapso y ulceración de un pólipo, obstrucción e invaginación intestinal son complicaciones de la enfermedad.2 El aumento de riesgo de presentar cáncer es a nivel gastrointestinal, mama, páncreas y órganos reproductivos (tumor de células de Sertoli, endocervical y tumor de ovarios).6
Es una enfermedad genética con herencia autosómica dominante (AD) y se caracteriza por una expresividad variable ya que algunos pacientes solo presentan hiperpigmentación (puede desaparecer con la edad), otros solamente pólipos, y otros manifiestan tanto hiperpigmentación como pólipos intestinales.3,7,8 Es causada por una mutación que produce pérdida de función en la línea germinal del gen supresor de tumores STK11 (seronine/threonine kinase) o LKB1 (liver kinase B1), el cual se localiza en el cromosoma 19p y codifica para una proteína serina/treonina quinasa (liver kinasaB1 [Lkb1]). El papel de este gen es actuar como un regulador negativo en varias vías metabólicas y su alteración conlleva una disminución en la inhibición del crecimiento y desarrollo celular obteniendo como resultado un crecimiento incontrolado de las células y de esta forma el desarrollo de poliposis intestinal y tumores.2,6,9
Entre el 94 y 96 % de los pacientes con SPJ se estima que tienen una mutación en STK11. Queda por determinar si un segundo gen de susceptibilidad en aquellos pacientes sin la mutación STK11 es responsable de la enfermedad.3,5 La penetrancia es completa, pues todos los individuos notificados con variantes patogénicas en STK11 han mostrado manifestaciones clínicas.3
El diagnóstico se basa en los hallazgos clínicos y la apariencia histológica de los pólipos que los distinguen de los pólipos del resto de los síndromes de poliposis hamartomatosos. La identificación de una variante heterocigótica en el gen STK11 mediante estudios moleculares confirma el diagnóstico.3,7
Las telangiectasias son dilataciones anormales de los vasos terminales del plexo subpapilar en la dermis superficial que son visibles como pequeñas lesiones de color rojo púrpura en la piel y las membranas mucosas.10,11 Pueden estar asociadas a enfermedades hereditarias o ser secundarias a enfermedades sistémicas.10
En la literatura se reporta un síndrome integrado por poliposis juvenil y telangiectasia hemorrágica hereditaria (THH).12 Por otra parte, también se ha descrito el prolapso de la válvula mitral junto a poliposis juvenil.13 Sin embargo, asociación entre SPJ, telangiectasias y prolapso de la válvula mitral en un mismo individuo no ha sido referido hasta ahora. Ambas asociaciones descritas anteriormente son atribuidas a mutaciones en el gen SMAD4 (SMAD family member 4).12,13
El objetivo de este trabajo es describir los hallazgos que permitieron establecer el diagnóstico de Síndrome de Peutz-Jeghers en un paciente, asociado a telangiectasias y prolapso de la válvula mitral, lo que nos permitió brindar asesoramiento genético a la familia.
Presentación de caso
Paciente masculino de 36 años de edad con antecedentes de prolapso de la válvula mitral que acude a consulta de genética clínica con su esposa para solicitar asesoramiento genético, debido a que tienen una hija con diagnóstico de Síndrome de Peutz-Jeghers y desean conocer el riesgo de tener otro hijo afectado. Como antecedentes familiares de interés al abuelo paterno del propósito se le diagnosticó de forma incidental un pólipo benigno en vías respiratorias a la edad de 58 años (no hay evidencia documental de su histología). Un tío y abuelo paterno son hipertensos y han presentado epistaxis ocasionalmente (Figura 1).14

Fig. 1 Arbol genealógico de la familia con SPJ. Números romanos indican las generaciones. Números arábigos indican los individuos de cada generación. Cuadrado = varón, círculo = hembra, asterisco= persona que solicita asesoramiento genético, lineas discontinuas= persona ausente. La flecha revela el caso índice o probando (individuo afectado a través del cual se identifica una familia con una condición genética, puede ser o no el individuo que solicita la consulta o el asesoramiento genético).14 Ambas barras oblicuas indican divorcio.
Al examen físico, la madre del propósito es aparentemente sana y en el padre se observa mácula hipermelánica en labio inferior, varias de estas en encías y se constatan múltiples telangiectasias en región de anterosuperior y posterosuperior del tórax y cuello, las cuales no habían sido diagnosticadas anteriormente (Figuras 1 y 2).
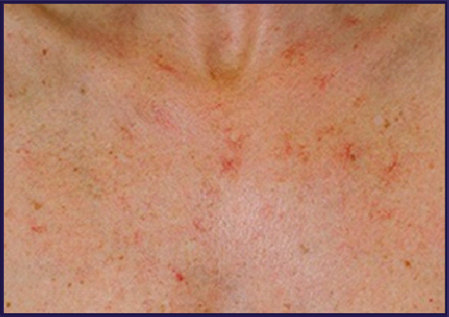
El paciente es valorado por la especialidad de gastroenterología. Se realiza endoscopia del tracto digestivo superior e ileocolonoscopia en busca de pólipos, las cuales fueron negativas; así como ultrasonido de abdomen superior donde se visualizó doble sistema pielocaliceal en ambos riñones. No fue factible llevar a cabo estudio endoscópico para visualizar intestino delgado. Los estudios de tipo analítico (hemograma, coagulograma, creatinina, glucosa, ácido úrico, proteínas totales, albúmina, colesterol, triglicéridos, ALAT, ASAT, GGT, globulina, amilasa y fosfatasa alcalina) resultaron normales. La carga viral para los virus B y C de la hepatitis fueron negativos.
Debido a la presencia de telangiectasias y los antecedentes familiares de epistaxis, el paciente es valorado por la especialidad de otorrinolaringología. El examen físico correspondiente fue negativo.
Discusión
La presencia de máculas melánicas en labio inferior y encías constatadas al examen físico del padre del probando (caso que se presenta) permitieron emitir el diagnóstico de SPJ, ya que uno de los criterios diagnósticos de esta entidad es la pigmentación mucocutánea característica en un individuo que tiene historia familiar de SPJ en al menos un pariente cercano.3 El estudio molecular no fue posible realizarlo. Si bien las máculas melánicas pueden formar parte de otras condiciones hereditarias como el complejo de Carney y el Síndrome LEOPARD,4 fueron descartadas ya que no se presentan en el caso en estudio los tumores endocrinos y dismorfias características de tales entidades respectivamente.15,16
Aunque este síndrome tiene una expresividad variable pudiendo la madre del probando presentar solamente pólipos hamartomatosos, hay que tener en cuenta que este trastorno es una entidad rara2,4 con herencia AD por lo que la probabilidad de que ocurra un emparejamiento entre dos personas afectadas de una población es muy poco probable. Sin embargo, la presencia de un pólipo benigno en vías respiratorias del padre del paciente pudiese ser una evidencia de que este individuo sea portador de SPJ y haber sido heredado el síndrome por vía paterna. Por los motivos anteriores, se decidió estudiar solamente al padre quien muy probablemente deberá ser heterocigótico (Aa), ya que los individuos homocigóticos dominantes (AA) son muy infrecuentes y estarán muy afectados como para reproducirse.
Debido a que no fue posible llevar a cabo estudio endoscópico para visualizar intestino delgado, la presencia de pólipos a este nivel no puede ser descartada. La edad de aparición de los síntomas relacionados con los pólipos es variable, y algunos individuos desarrollan síntomas durante los primeros años de vida.3 En un estudio realizado en España por Rodríguez y colaboradores, el diagnóstico fue realizado entre uno y 32 años, y la edad media fue de 13,8.2) El caso que se presenta tiene 36 años de edad y hasta este momento no había sido diagnosticado. Otro elemento a tener en cuenta es la expresividad variable que caracteriza a este síndrome, por lo que este paciente pudiera presentar solamente las máculas hipermelánicas descritas al examen físico.
En relación con las telangiectasias descritas en el paciente se presenta la interrogante de si existe una relación entre estas y el SPJ, o si es solo una cuestión de coincidencia. Elementos como la edad de aparición de las telangiectasias, si otros miembros de la familia son portadores de estas, así como el patrón de distribución y la presencia de otras características son importantes para responder la interrogante. No fue posible obtener la información de los dos primeros.
Las telangiectasias pueden ser primarias como una característica que acompaña a enfermedades hereditarias entre las que se encuentran la ataxia- telangiectasia, xeroderma pigmentoso, síndrome de Bloom, telangiectasia hemorrágica hereditaria (THH) o Síndrome Rendu-Osler-Weber, telangiectasia hereditaria benigna, entre otras; y secundarias en el curso de enfermedades sistémicas como cirrosis hepática, vasculopatía del colágeno, dermatomiositis, telangiectasia macular, tumores metastásicos, o como resultado de factores externos (medicación, radioterapia, exposición al sol y traumatismo).11,17 No hay antecedentes de estos últimos en la historia del paciente.
El hecho de que las telangiectasias se distribuyeran a nivel de la región superior del tórax y cuello en el caso en estudio, no estar asociadas a algún otro signo o síntoma, no haberse encontrado alteraciones en órganos intrabadominales ecográficamente, y la analítica estar dentro de límites normales, pudiera corresponderse con el diagnóstico de una telangiectasia hereditaria benigna. Este es un trastorno AD que cursa con múltiples telangiectasias cutáneas, aparecen en los primeros años de vida y se limitan a las zonas expuestas al sol, o pueden distribuirse aleatoriamente por el cuerpo. Debe sospecharse en personas sin antecedentes personales y/o familiares de hemorragias y/o epistaxis, telangiectasias mucosas y malformaciones arteriovenosas.11,12
No obstante, un dato a tener en cuenta es que el padre y el hermano del paciente por vía paterna tienen antecedentes de epistaxis recurrentes e hipertensión arterial (Figura 1). Una posible asociación entre ambas entidades ha sido reportada en algunos estudios,18 aunque en este caso se desconoce si estas han ocurrido simultáneamente en estos individuos. Aunque las telangiectasias y epistaxis pueden observarse en personas sanas, ambos signos forman parte del síndrome de THH.12
Este diagnóstico se establece en un individuo con tres o más de las siguientes características clínicas: epistaxis, telangiectasias mucocutáneas, malformaciones arteriovenosas viscerales y/o antecedentes familiares. La identificación de una mutación germinal en uno de los genes ACVRL1, ENG, GDF2 y SMAD4 determina el diagnóstico si las características clínicas no son concluyentes.12
Debido a que se desconoce si los familiares referidos anteriormente presentan o no telangiectasias u otros elementos propios del síndrome, no es posible establecer el diagnóstico. Por otra parte, esta condición presenta gran variabilidad intrafamiliar y una penetrancia relacionada con la edad lo que significa que las manifestaciones se desarrollan a lo largo de la vida. Hasta el 95 % de los individuos afectados eventualmente experimentan epistaxis recurrente, un tercio comienza a los diez años, aproximadamente el 80 % a los 20 años y el 90 % antes de los 30 años.12¿Pudiera el paciente en estudio desarrollar epistaxis a edades más tardías?
La edad de inicio de las telangiectasias es generalmente más tardía que las epistaxis pero puede ser durante la infancia.12 Aproximadamente 25 % de los individuos con THH presentan hemorragia por telangiectasias gastrointestinales y por lo general el sangramiento comienza después de los 50 años. Este se presenta con más frecuencia como anemia ferropénica que como sangramiento digestivo agudo.12 El caso que se presenta tiene 36 años, no presentó anemia al momento del estudio y hasta donde permitió observar el examen endoscópico no se encontraron telangiectasias.
Las malformaciones arteriovenosas se manifiestan con más frecuencia en hígado, cerebro y pulmón, pero pueden no siempre estar presentes.12 No han sido realizados estudios para su detección en el paciente.
Se han informado mutaciones en el gen SMAD4 en familias con un síndrome combinado de poliposis juvenil y THH;19 sin embargo, la asociación entre SPJ y telangiectasias aún no ha sido reportada en la literatura. Sweet y colaboradores encontraron en un individuo diagnosticado con poliposis juvenil una deleción de 29 pares de bases del exón 1 de STK11 y un polimorfismo en IVS7-32 (A → T) en el gen SMAD4.20
Por otra `parte, no todos los pacientes con diagnóstico de SPJ presentan mutación en el gen STK11.2 Andrabi y colaboradores reportaron un paciente con poliposis juvenil y prolapso de la válvula mitral asociado a mutación en el gen SMAD4. Este gen actúa en la vía de señalización del factor beta de crecimiento transformante (TGFB) y tiene doble función como regulador transcripcional y como supresor de tumores dentro de esta vía. Se ha demostrado que el aumento de la señalización del TGFB puede contribuir a cambios mixomatosos de la región mitral.13
Se ha señalado que existe una relación funcional entre AMPK (de sus siglas en inglés AMP-activated protein kinase), un importante efector en dirección 3̕ de STK11 con función preventiva e inhibidora sobre la tumorgénesis, y el TGFB. La expresión de STK11 impide la fosforilación de SMAD inducida por TGFB y su actividad transcripcional, mientras que la eliminación de STK11 potencia el efecto de TGFB. Por tanto, si el paciente en estudio es portador de SPJ y fuese debido a una mutación en STK11 que produce pérdida de función en la proteína que codifica, esto potenciaría el efecto de TGFB y contribuiría al prolapso de la válvula mitral que presenta.21,22
Todo el análisis anterior sugiere, que a pesar de que el diagnóstico de SPJ fue confirmado por los hallazgos clínicos, en opinión de los autores, sería importante en este paciente realizar el estudio molecular con el objetivo de buscar mutaciones tanto en el gen STK11 como en otros genes relacionados con otros síndromes hamartomatosos. De esta manera se podría establecer una vigilancia, así como, trazar estrategias de intervención para el paciente y los familiares en riesgo cuando no es posible determinar el origen de signos que no forman parte del SPJ (telangiectasias y prolapso de la mitral), teniendo en cuenta que ha sido reportado en la literatura la asociación de otro síndrome hamartomatoso (poliposis juvenil).
En relación con el asesoramiento genético, la descendencia de un individuo con SPJ tiene un 50 % de probabilidad de heredar la enfermedad. Se proporcionará al paciente la posibilidad de realizar de forma periódica estudio endoscópico de vías digestivas, con el objetivo de detectar y tratar precozmente la presencia de pólipos, así como, ecografía testicular y de páncreas para la vigilancia de cáncer. Dado que se desconoce la mutación causante del SPJ se ofrecerán evaluaciones clínicas a familiares de primer grado (padre y hermanos del paciente), quienes se beneficiarán de un tratamiento temprano y una vigilancia adecuada. En caso de tener otro hijo se brindará la opción de estudio endoscópico a partir de los ocho años si es asintomático, y más tempranamente si es sintomático.4 Hay que tener en cuenta que el padre del paciente tiene antecedentes de pólipo en vías respiratorias lo cual puede formar parte del cuadro de SPJ.
Conclusiones
Los antecedentes y los hallazgos encontrados en el paciente permitieron realizar el diagnóstico de Peutz-Jeghers y brindar un asesoramiento genético. Se presenta el primer reporte en la literatura científica en el cual se describe de manera conjunta SPJ, telangiectasias y prolapso de la válvula mitral.

