










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkIngeniería Industrial
versión On-line ISSN 1815-5936
Ing. Ind. vol.33 no.1 La Habana ene.-br. 2012
ARTÍCULO ORIGINAL
Liberación de lotes para productos en desarrollo
Batch release for investigational products
Daymara González-Fuentes, I Mercedes Delgado-Fernández, II Lizette Fontanet-Tamayo, I Antonio Vallín-García, I
I Centro de Inmunología Molecular (CIM). La Habana, Cuba.
II Instituto Superior Politécnico José Antonio Echeverría, Cujae. Facultad de Ingeniería Industrial. La Habana, Cuba.
RESUMEN
Este artículo describe un procedimiento de liberación de productos en desarrollo del Centro de Inmunología Molecular. El Procedimiento incorpora el conocimiento y la complejidad de la investigación y desarrollo (I+D), análisis de riesgo, correlaciones y análisis factorial; evaluando parámetros clínicos, características de calidad y controles de procesos, para asegurar la calidad del producto. Con la aplicación sistemática del nuevo procedimiento, se disminuirá el tiempo de liberación significativamente, garantizando la calidad del producto, la ejecución de las regulaciones, el espacio de diseño y un mayor conocimiento de los procesos y productos en desarrollo.
Palabras clave: liberación de lotes, calidad en el desarrollo de productos, espacio de diseño, correlaciones, análisis factorial.
ABSTRACT
This paper describes a procedure for the release of investigational products at the Center of Molecular Immunology. The Procedure incorporates the knowledge and the complexity of R&D, risk assessment, correlations and factorial analysis, evaluating clinical parameters, characteristic of quality and process controls to assure the quality of the product. The systematic application of the new procedure will diminish the release time significantly, guaranteeing the quality of the product, the compliance with national/international regulations, the design space and a greater knowledge of the processes and products in development.
Key words: batch release, quality in product development, design space, correlations, factorial analysis.
INTRODUCCIÓN
Gestionar la calidad en la industria biofarmacéutica constituye un gran reto, sobre todo si se trata de insertarla en cada una de las etapas por las que atraviesan dichos productos [1], dentro de las cuales se encuentra la etapa de investigación y desarrollo (I+D). La I+D tiene un papel vital en la industria biofarmacéutica y en ella se concentran los mayores esfuerzos para la transformación de los resultados científicos en productos comerciales [2], destinando como promedio el 45 % de sus ganancias anuales a esta etapa, con la protección por patentes para prácticamente todos los productos [3].
La I+D comienza en la investigación básica, la cual demuestra con estudios preclínicos, la eficacia del producto sobre un blanco determinado y su acción principal; y continúa con el desarrollo del producto, donde se definen funcionalmente 3 fases de ensayos clínicos, cada una de las cuales es diseñada para responder a diferentes objetivos en la investigación [4]. Esta etapa también incluye el escalado, aunque la fase clínica determina aún el desempeño de un producto (cerca del 80 % del gasto y del tiempo es debido al desarrollo clínico). El concepto de desarrollo actual "asume" que el paso limitante del desarrollo es la etapa clínica. En el centro donde se ha llevado a cabo esta experiencia durante más de 10 años, el escalado no constituye el problema fundamental. Este artículo ofrece una alternativa para que el escalado y desarrollo del proceso y producto sea más expedito, al definir claramente cuáles son los atributos críticos del producto y los que tienen que permanecer "constantes" a lo largo del desarrollo.
El desarrollo comienza con la fase I del ensayo clínico, para chequear la seguridad y la dosis óptima del medicamento; la fase II, para comprobar su eficacia y efectos colaterales; la fase III, para monitorear las acciones del medicamento en un largo tiempo de uso; y concluye con la revisión y aprobación de un Registro del producto por una agencia reguladora. Se evidencia un cambio de concepto en las regulaciones de los medicamentos biológicos (al menos los recombinantes), al realizarse las fases II con ensayos clínicos controlados y a dobles ciegas. Muchos de estos ensayos son aceptados como registros condicionados para indicaciones con escasas alternativas terapéuticas. Las fases III se convierten en ensayos confirmatorios, lo cual es especialmente cierto en los productos para el tratamiento del cáncer, que son el objeto de este artículo. Cada vez más conducir ensayos fases III en cáncer, a doble ciegas, con cientos de pacientes, constituye un problema ético. Cuando existen productos biosimilares, las nuevas regulaciones permiten tomar la data precedente y realizar análisis con la misma para extrapolar resultados, por lo que, la agencia reguladora permite no realizar determinadas evaluaciones y por eso las fases parecen ir más rápido.
La I+D transita hasta obtener finalmente un proceso productivo capaz de satisfacer los requerimientos para los cuales fue diseñado. Mientras más pronto se logre disponer (registrar) de un producto seguro, eficaz y sobre todo novedoso, más rápido se recuperará la organización de los gastos en que incurrió durante su I+D. Se plantea que estos productos en desarrollo pudieran representar en el mercado mundial dividendos potenciales en el orden de los 20 billones de dólares. Un objetivo fundamental es hacer cada vez más corta esta etapa I+D y que los productos lleguen a los pacientes que lo necesitan con la rapidez y calidad necesaria.
Actualmente las autoridades flexibilizan las regulaciones y los mecanismos de desarrollo de producto, creando así un marco más favorable para la entrada de productos en el mercado, con esfuerzos de armonización en los sistemas de calidad [5], en las Buenas Prácticas de Fabricación (BPF), calidad en el diseño [6] y análisis de riesgos [7]. Es importante puntualizar que el concepto de flexibilizar no quiere decir relajar, sino que permiten un enfoque de gestión del conocimiento, que con anterioridad a estas nuevas regulaciones no era posible plantear. El manejo de riesgo y de la posibilidad de tener "procesos y moléculas definibles", es una muestra de madurez de la industria. El concepto de criticidad quiere decir que no todo es igualmente importante. A esto se refiere la "flexibilidad".
El Centro de Inmunología Molecular cuenta con una carpeta de más de 20 productos en I+D, por lo que diseñar e implementar nuevas funciones basadas en estos principios de las nuevas regulaciones, posibilita la disminución del tiempo de liberación de lotes de productos en desarrollo, teniendo en cuenta la integración del conocimiento científico, los requerimientos regulatorios y el análisis de riesgo [8]. Esto permite, además, la disminución de no conformidades detectadas por las agencias regulatorias en el momento de solicitar el registro, para que este trámite ocurra lo más pronto posible y en la mayor cantidad de países. En este artículo se describe un procedimiento de liberación de lotes para productos en desarrollo, diferente al ya existente para los productos comerciales, así como una instructiva para el análisis de riesgo, que permite la liberación de estos productos enfocado al espacio de diseño. Se realizan, además, correlaciones entre parámetros clínicos, de proceso y atributos de calidad y el análisis multivariado para un producto del Centro, que permite evaluar la criticidad de los atributos de calidad y obtener mayor conocimiento del proceso y producto.
MÉTODOS
Este acápite se estructura en 2 partes: una relacionada con el enfoque y el concepto referido al espacio de diseño, que se ha establecido para el desarrollo de productos farmacéuticos; y la otra parte relacionada con la propuesta del procedimiento para la liberación de lotes en la fase de desarrollo de productos biofarmacéuticos.
Espacio de diseño
El concepto de «espacio de diseño» exige que un producto biotecnológico sea diseñado para que alcance su rendimiento clínico deseado y que el proceso sea capaz de ofrecer consistentemente un producto que cumple con los atributos de calidad necesarios para dicha acción clínica [9]. La ventaja del espacio de diseño es la flexibilidad regulatoria, por la posibilidad de realizar cambios en los procesos dentro del espacio de diseño sin la autorización de la agencia reguladora. Para alcanzar el nivel requerido de conocimiento del proceso, son necesarios estudios de caracterización con una amplia gama de parámetros de proceso y con la variabilidad aceptable [10]. Los atributos de calidad se establecen en base a la clínica del producto, el conocimiento de otros productos similares y la comprensión científica general de la molécula. Los estudios de caracterización se realizan para establecer rangos de intervalos aceptables a los parámetros operativos claves y críticos. Operando dentro de estos rangos aceptables, la combinación de lo que finalmente define el espacio de diseño, ofrece la garantía de calidad [11]. Un restablecimiento del espacio de diseño inicial para la fermentación, purificación y llenado de productos, debe tener en cuenta un mayor conocimiento del proceso y del producto a través de ensayos clínicos y no clínicos, estudios de estabilidad, cambios de proceso, ejercicios de comparabilidad, tecnologías de plataforma y nuevas tecnologías analíticas.
El restablecimiento del espacio de diseño se basa en los mismos principios utilizados para desarrollar el espacio de diseño inicial. A partir del perfil de calidad de los productos, se debe realizar una evaluación del riesgo [12] para identificar los parámetros del proceso que serán reevaluados con respecto a su posible impacto sobre la calidad de los atributos críticos, usando modelos de pequeña escala y la aplicación del diseño de experimentos [10].
Al tener rangos amplios en los parámetros que se miden, más extenso será también el espacio de diseño y existirá mayor flexibilidad en la producción y transferencias, lo cual permitirá caracterizar mejor un proceso y un producto y, además, establecer especificaciones basadas en el conocimiento. Una vez definido el espacio de diseño para un producto o proceso y aprobado por la agencia reguladora, cualquier variación dentro de éste, no constituye un cambio. La calidad por diseño (CpD) es una parte esencial del enfoque moderno de la calidad farmacéutica. Constituye el enfoque sistemático para el desarrollo farmacéutico, enfatizando en la comprensión de cómo pueden influir los parámetros de proceso sobre las variables de calidad de los productos y en la gestión del riesgo [13].
El espacio de diseño queda definido al concluir la fase III de desarrollo del producto, en la cual ya estarán definidas las especificaciones de calidad [14], basado en la caracterización y estudios de formulación, que contribuirá a la selección de atributos de calidad que garanticen que su cumplimiento en cada lote conlleve a una respuesta segura y eficaz en los pacientes.
Durante la fase de desarrollo se identificarán los parámetros críticos y métodos que se utilizarán para controlarlos durante el proceso. Los parámetros de producción y los controles provisionales durante el proceso, generalmente pueden ser deducidos de la experiencia previa, incluyendo la adquirida en las tempranas etapas de desarrollo y con productos análogos.
Seguir las nuevas guías armonizadas para la I+D en la industria biotecnológica y tomar determinados conceptos básicos como el de calidad por diseño, es hoy un reto para el Centro de Inmunología Molecular, ya que éste cuenta con varios productos en esta etapa y debe alcanzar el mayor grado de conocimiento científico, capaz de satisfacer las exigencias requeridas para un producto de esta categoría. Una forma de reducir este tiempo y aprovechar cada vez más la patente de un producto, es mediante la reducción del tiempo de liberación de los lotes de productos en desarrollo que van a ser usados en los ensayos clínicos, según el espacio de diseño.
Procedimiento de Liberación de lotes pata productos en desarrollo
El proceso de liberación de lotes para productos en desarrollo que se lleva a cabo en el Centro de Inmunología Molecular, tiene la finalidad básica de liberar los lotes de los productos de la Planta Piloto, para su uso en ensayos clínicos en sus respectivas fases. Con anterioridad se aplicaba el mismo procedimiento en el desarrollo de productos, que el empleado en los productos comerciales, sin diferenciar las fases I, II y III de ensayos clínicos.
Las actividades a realizar por los especialistas del grupo de liberación de lotes del Departamento de Aseguramiento de la calidad para productos en desarrollo son:
- Revisión de la documentación emitida por los productores.
- Verificación del cumplimiento o no de los parámetros de control de la calidad y en el caso que se requiera, realización de un análisis de riesgo.
- Emisión de la documentación correspondiente a cada lote (certificado y registros correspondientes).
- Conformación del expediente del lote.
Para la revisión de la documentación emitida por los productores, se tendrá en cuenta la presencia y vigencia de los registros que conforman el expediente de lote de cada producto. Las materias primas utilizadas estarán liberadas y dentro de su período de vigencia, según requerimientos del Procedimiento Normalizado de Operación (PNO) de liberación de materias primas de productos en desarrollo. Se cumplirán las instrucciones de trabajo establecidas para cada etapa del proceso productivo y de no ser así, todos los cambios serán documentados en los registros correspondientes. También se cumplirá lo establecido en las Buenas Prácticas de Producción con respecto a la documentación de los reportes, tales como: los cromatogramas, registros de temperaturas, ciclos de esterilización y pruebas de integridad. Las soluciones empleadas cumplirán con sus especificaciones y serán usadas dentro de su fecha de vigencia. El material empleado será el adecuado y se encontrará dentro del tiempo de vigencia de preparación. Los límites microbiológicos establecidos durante el proceso de producción y llenado y la calidad del agua de cada etapa serán chequeados, emitiéndose la conformidad de dichos parámetros en un informe.
Para la verificación del cumplimiento de los parámetros analizados por Control de la Calidad (CC), se tendrán en cuenta las fases de ensayo clínico en que se encuentra el producto y se llevará a cabo un análisis de riesgos, si existiese algún ensayo fuera de especificación. Esto significa que un producto que se encuentre en fase I de desarrollo, no podrá tener el mismo análisis que uno que se encuentre en fase II o III; ya que en fase I, el producto se encuentra comenzando su desarrollo y aún no se conoce mucho de éste. De hecho, se propone que al inicio de la etapa de desarrollo se prueben varios métodos de potencia, ya que en ese momento el mecanismo de acción de la molécula no es totalmente conocido y esto permitirá incorporar el conocimiento útil que lo vaya definiendo. Además, se propone que se mida la mayor cantidad de variables posibles que ayuden a la caracterización de los productos. En estas etapas tempranas las fichas de especificación emitidas tendrán límites de aceptación lo más amplios posible, bien justificados y documentados por los investigadores y productores, que permitan tener elementos para tomar decisiones en el momento de la liberación de un lote al manejar el riesgo.
En etapas más avanzadas se cuenta con mayor cantidad de datos, porque al concluirse el primer ensayo clínico, al menos se tienen datos de seguridad, dosis máxima tolerable y de caracterización, que van ayudando a conformar el espacio de diseño, por lo que los rangos de aceptación deben ir estrechándose y apuntar cada vez más al rango óptimo. Así, se podrá analizar el cumplimiento de los rangos de aceptación establecidos y en caso de no cumplirse con esos rangos, se analizará el riesgo según la instructiva establecida para este tipo de análisis. Ese análisis de riesgo plantea que si la desviación que se presenta en el lote está dentro del espacio de diseño que se ha ido conformando para el producto, esto no constituye un cambio, pues ya se ha demostrado en etapas anteriores que aunque no es la condición óptima, funciona dentro de los límites aceptables y por tanto, se acepta el lote y no se tiene que recurrir a ninguna aprobación externa.
Si se cumplen todos los requisitos anteriormente descritos, se procede a la liberación del lote, como se representa en la figura 1. En todas las fases de desarrollo se tendrá en cuenta la flexibilidad que brindan las nuevas guías, haciendo hincapié en las guías ICH Q8 e ICH Q9, donde se ponen de manifiesto dos conceptos fundamentales: espacio de diseño y análisis de riesgo.
La instructiva de trabajo para el análisis de riesgo, que se muestra en la figura 2, se aplica a todos los productos que se encuentren en las fases de desarrollo (incluye las materias primas, disolventes, excipientes, envasado y etiquetado de las materias en las drogas).
La valoración del riesgo para la calidad se basa en conocimientos científicos y según se establece en la instructiva correspondiente, el especialista debe hacer primeramente una identificación del riesgo, donde a través de datos históricos, moléculas o procesos similares, se puede conocer la existencia de riesgos potenciales y sus posibles consecuencias, lo cual conlleva a la toma de acciones preventivas y un estricto control. En base a esto se analiza el riesgo, teniendo en cuenta su probabilidad de ocurrencia y gravedad de los daños que se pueden ocasionar. En este momento se valora incluso la capacidad de control que se tiene sobre ese riesgo. Esa información permite estimar cuantitativamente el riesgo y clasificarlo como sigue:
- Riesgo alto: Representa un impacto significativo inmediato o latente para la salud. Puede influir negativamente en la calidad, seguridad o eficacia de los medicamentos.
- Riesgo mediano: Aquel medicamento producido que no cumpla con la calidad requerida para el ensayo clínico (consistente y adecuado a la fase en que se encuentre).
- Riesgo bajo: Aquel que no se encuentra en las clasificaciones antes mencionadas, pero que incurre en un incumplimiento de las buenas prácticas en general.
Si se acepta el riesgo, se liberará el lote, emitiéndose el certificado de liberación final y, en caso contrario, se rechazará el lote.
Para el registro y procesamiento de los datos se usan herramientas estadísticas para la evaluación efectiva y la gestión del riesgo, tales como: gráficos de control, diseño de experimentos, histogramas, diagramas Pareto, análisis de capacidad de proceso, correlación y análisis factorial.
El análisis de correlaciones se puede aplicar a los datos de controles de proceso, clínicos y atributos de calidad, que permitirán potencialmente obtener mejores resultados en la clínica, variando algunos parámetros de proceso según las relaciones entre estas variables. Así, se conoce más de los productos en desarrollo y su comportamiento en la clínica, integrando el conocimiento científico con los requisitos regulatorios y estableciendo las especificaciones de los productos según su funcionamiento. La actividad biológica medida a un producto, debe ser medida fiel de la respuesta clínica de éste.
También se elaboraron listas de chequeo en las que se establecen los estudios a realizar para cada fase de ensayo clínico (I, II y III). La tabla 1 muestra 2 de los aspectos a completar de las investigaciones no clínicas: farmacología y toxicología.
RESULTADOS
Como aplicación del espacio de diseño se estudió la vacuna 1E del CIM, que se encuentra en fase II de desarrollo y para ello se hizo un análisis retrospectivo de sus datos clínicos y de proceso, obteniéndose conclusiones muy importantes.
Este candidato vacunal está compuesto por el anticuerpo monoclonal (AcM) murino 1E10, que mimifica el gangliósido NGcGM3, antígeno expresado en algunos tumores humanos, como por ejemplo, tumores de mama y pulmón; por lo que su administración como vacuna debe levantar una respuesta inmune específica contra células tumorales que presenten este antígeno de superficie.
El proceso productivo de este AcM comienza con la inoculación de los hibridomas que producen esta proteína en ratones para obtener líquido ascítico murino (LAM). Posteriormente la purificación de este AcM se realiza mediante 3 pasos cromatográficos: intercambio iónico (DEAE), afinidad (Proteína A) y exclusión molecular (G25). Finalmente en el paso de formulación, este AcM se conjuga con el adyuvante hidróxido de aluminio para formar la vacuna 1E10/ alúmina.
Para realizar este análisis, se usaron los datos de los parámetros de laboratorio clínico medidos para los pacientes de un ensayo clínico Fase II con la vacuna 1E y los expedientes de los lotes usados para la vacunación de los mismos, calculándose las correlaciones.
Se tomaron como parámetros de proceso la masa de IgG que entra a la columna de purificación y que fue medida en el LAM, el volumen de anticuerpos obtenido en la purificación, así como la masa de proteínas totales, la concentración de las mismas y rendimiento total del proceso. Los atributos de calidad seleccionados fueron la actividad biológica, pH y pureza cuantitativa del ingrediente farmacéutico activo (IFA); y del producto final, se tomaron la actividad biológica, pH, concentración de 1E10 y % de adsorción. Es válido aclarar que aunque la concentración de ión Al+++ final no era un ensayo a medir para la liberación del lote, sí fue evaluado en cada uno para la posterior determinación de su intervalo de aceptación, y por tal motivo se contó con los resultados de este parámetro, de gran importancia para este trabajo, debido a su actividad adyuvante. Finalmente, comoparámetros clínicos se tuvieron en cuenta la sobrevida de los pacientes, título máximo alcanzado contra 1E10 y título máximo de IgG e IgM contra gangliósidos.
Se realizaron correlaciones entre todas estas variables y entre las más significativas se encuentra la correspondiente al título máximo de Ac contra 1E10 alcanzado por las pacientes y el título máximo de IgG contra gangliósidos también alcanzado por las pacientes (coeficiente de correlación (r) = 0,785 y significación estadística (p) = 0,000. Se corrobora que la respuesta contra 1E10 influye en la respuesta contra gangliósidos y que el idiotipo es lo que mimifica el gangliósido.
A su vez se observa una correlación negativa significativa entre las concentraciones de Al+++ y estos 2 parámetros clínicos (Ac contra 1E10 ((r) = -0,367 y (p) = 0,002) y Ac contra gangliósidos ((r) = -0,318 y (p) = 0,008)); es decir, mientras aumenta uno, disminuye el otro. Para el análisis de esta correlación fue necesario tener en cuenta la correlación positiva y significativa que existe entre la concentración de Al+++ y el % de adsorción, medido al producto final ((r) = 0,528 y (p) = 0,000), ya que existen estudios anteriores en los que la mayor adsorción del Al+++ propicia mayor respuesta inmunogénica del producto en los pacientes por la capacidad adyuvante de la alúmina. No obstante, estudios recientes apuntan a que en el caso del 1E10, esta relación se debe interpretar de otra forma y esta correlación negativa anterior es un ejemplo de ello, lo que puede llevar a pensar que mayor cantidad de aluminio y por tanto mayor % de adsorción implica menores títulos de Ac contra 1E10 y contra gangliósidos. De ahí que se debe analizar el mecanismo de acción de esta molécula y el papel adyuvante de la alúmina en la misma.
Otra variable con correlaciones significativas positivas entre los parámetros título contra 1E10 y título contra gangliósidos, fue con la pureza cuantitativa que se mide en el ingrediente farmacéutico activo. Esta variable mide el porcentaje de la molécula que se encuentra en forma de monómeros y dímeros, y aunque aún no se conoce qué sucede con dicho porcentaje después del paso de formulación. Es un dato de interés saber que mientras mayor es esta variable en el IFA (ya que en el producto final no se puede medir porque ya se encuentra conjugada la molécula con la alúmina), mayor es el título de los pacientes contra 1E10 y contra gangliósidos.
El análisis de la actividad biológica del producto final y la realizada a los lotes de IFA correspondientes, arroja una correlación negativa significativa ((r) = -0,348 y (p) = 0,003). Aunque la actividad biológica final tuvo correlaciones positivas significativas con la concentración de Al (r = 0,593 y p = 0,000) y con el % de adsorción (r = 0,449 y p= 0,000); esta correlación aún no tiene un fundamento científico que pueda corroborarlo.
Se confirmó además, la correlación positiva significativa, descrita en la literatura, entre la concentración de Al y el porcentaje de adsorción (r = 0,528 y p = 0,000).
Otro parámetro que muestra correlación significativa positiva es la actividad biológica final y el pH (r = 0,857 y p= 0,000). Los pH cercanos a 7,5 (límite superior de la especificación para el producto) facilitan el reconocimiento al blanco, lo cual coincide con el pH fisiológico, que es donde se favorecen las interacciones ligando-receptor o antígeno-anticuerpo.
Algo muy importante a destacar en este análisis, fue la no correlación de los parámetros medidos a los pacientes con la actividad biológica de los lotes de producto final, lo cual hace pensar que el método de evaluación de la actividad biológica utilizado para la liberación de los lotes de IFA y del producto final, no es indicativo de la eficacia de este producto. Esto exige el diseño e implementación de un método eficiente de actividad biológica, que se convierta en un verdadero atributo crítico de calidad que mida la eficacia de este preparado vacunal, previo al comienzo de los estudios clínicos fase III, en concordancia con las nuevas tendencias regulatorias para los productos en I+D.
Finalmente, se tienen las correlaciones encontradas dentro de las operaciones unitarias como la purificación. Mejorar los rendimientos de cada proceso para que constituyan una buena variable de entrada para el siguiente paso de producción, es algo que se debe hacer cada vez que sea posible, en correspondencia con la mejora continua de todo sistema de gestión de la calidad. En este caso se obtuvo una correlación negativa significativa entre la cantidad de IgG que entra al proceso de purificación y el rendimiento total de dicho proceso (r = -0,860 y p = 0,000). Existen diversas interpretaciones de esta situación. La primera sería analizar en qué parte de la campana están trabajando las columnas de afinidad que se usan para este proceso, según la saturación de las mismas. Otra variante analizada fue la capacidad de las columnas usadas, donde las mismas admitan determinada cantidad de producto a purificar y cómo, cuando se añade más de esa cantidad prevista, se mantiene bajo el rendimiento. Es decir, este paso requiere de un análisis que quizás ayude a mejorar su rendimiento.
Todo lo antes expuesto fue soportado en hipótesis a las se arribaron en reuniones con especialistas, que aunque no son resultados definitorios, pueden tener un basamento científico según los resultados obtenidos y ayudan a comprender la acción de los productos y a guiar los nuevos experimentos a realizar.
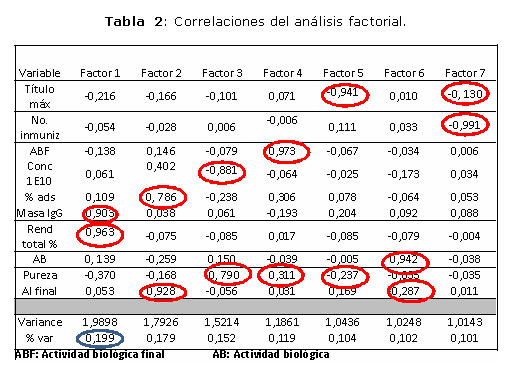
Otra herramienta posible a aplicar en el espacio de diseño es el análisis factorial [15]. Éste se realizó con el objetivo de analizar las interdependencias entre las variables del análisis de correlaciones. Se seleccionaron las variables que tuvieron un impacto en dicho estudio. La información que se observa a continuación en la tabla 2, muestra los resultados del análisis factorial con la rotación Varimax sólo para aquellos índices mayores que 1.
Con la aplicación del nuevo PNO de liberación de lotes de productos en desarrollo, se pudo disminuir el tiempo de liberación, al establecerse un tratamiento diferente para los productos en desarrollo según la fase en que se encuentran y el rigor con que deben ser analizados, además de tenerse un mejor conocimiento de los productos y procesos.
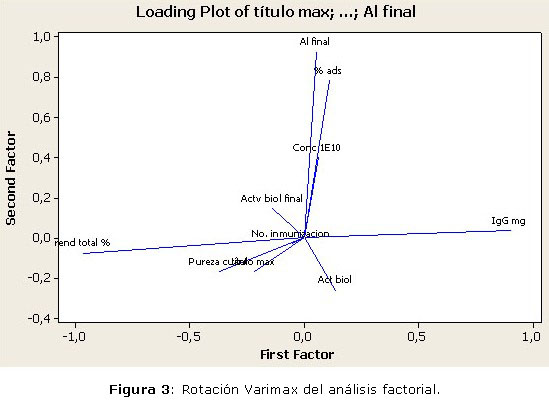
Se circulan las variables que mejor están interpretadas en cada factor, siendo el valor el coeficiente de correlación lineal con cada factor, por lo que se puede inferir cuáles son las variables que más están asociadas para cada factor. En el caso del factor 1, las variables mejor expresadas son la IgG, en sentido opuesto al porcentaje de rendimiento total. La varianza que se representa en este factor es del 19,9 %, siendo la del segundo factor 17,9 %. En el segundo factor, las variables que mejor lo expresan son el porcentaje de adsorción y el aluminio final, ambos en el mismo sentido de asociación. La figura 3 muestra el análisis realizado anteriormente. El resto de los factores se interpretan de la misma forma, para lo cual las correlaciones circuladas en cada factor son las que deben ser tenidas en cuenta para las interpretaciones del significado de cada factor.
DISCUSIÓN
Los resultados expuestos en el capítulo anterior son sólo un pequeño ejemplo del conocimiento que se puede adquirir en la etapa de desarrollo de un producto. Realizar este tipo de ejercicio complementa los experimentos que se ejecutan e incluso, pueden ser una hipótesis a comprobar en muchos casos.
El enfoque de calidad por diseño está descrito para ser implementado desde las fases tempranas de desarrollo, pero realizar este tipo de análisis retrospectivo permite aplicar principios importantes a aquellos productos que no tuvieron en su inicio de desarrollo este enfoque, porque aún no existían esas guías Q8, Q9 y Q10, permitiendo así establecer una relación entre los atributos de calidad y la respuesta clínica de los pacientes. Es decir, este estudio permite evaluar la criticidad de los atributos de calidad, según su impacto en la seguridad y eficacia del producto en los pacientes y a su vez, establecer qué parámetros de proceso influyen en esos atributos críticos. En esta relación se basan las nuevas guías ICH que permiten orientar el desarrollo de un producto a aquello que es realmente importante y crítico, es decir, al paciente.
Por tanto, esos atributos que tiene una correlación significativa con la respuesta de los pacientes, deben ser bien controlados o, al menos, bien estudiados, para evitar afectar su respuesta en la clínica; son parámetros que deben tener muy bien justificados sus rangos de especificaciones y una vez que sean aceptados por la agencia reguladora, para ser cambiados deben tener la aprobación de la misma. De igual forma, se deben tratar aquellos parámetros de procesos que tienen una correlación significativa con los atributos críticos de calidad.
Las nuevas guías, además, plantean que el enfoque tradicional y el de calidad por diseño no son mutuamente excluyentes. En el caso del CIM, este es el principio que se sigue, porque se encuentra en un período de transición donde existen productos en etapas avanzadas de desarrollo de los cuales se puede extraer conocimiento y este trabajo es un ejemplo de ello.
CONCLUSIONES
El Procedimiento de Liberación de Lotes para el desarrollo de productos en el Centro de Inmunología Molecular, utilizando las correlaciones entre los parámetros de proceso, especificaciones de producto y parámetros clínicos, facilita la evaluación de la criticidad de los atributos, la gestión de riesgos y el concepto del espacio de diseño.
La aplicación del espacio de diseño, mediante la caracterización de los parámetros de proceso y clínicos y especificaciones del producto con el análisis de correlación y el análisis factorial en un caso de estudio, permite demostrar la factibilidad y utilidad de este enfoque, al disminuir el tiempo de liberación de los lotes, acercar al centro a las actuales tendencias regulatorias armonizadas de las ICH Q8 e ICH Q9, así como ganar en conocimiento sobre el proceso y el producto.
El beneficio que reporta la aplicación del procedimiento de liberación de lotes en desarrollo es mucho mayor con respecto a los costos, teniendo en cuenta que influirá en que mayor número de pacientes se beneficien con el producto, se obtengan mayores ganancias en términos económicos y un mayor prestigio del Sistema de Salud e Industria Biofarmacéutica Cubana.
Se recomienda se generalice el procedimiento a otros centros biofarmacéuticos cubanos, que se divulguen los resultados obtenidos con la aplicación sistemática de dicho procedimiento en el CIM, sirviendo también de base para la mejora continua del mismo.
REFERENCIAS
1. LAGE, A., «La economía del conocimiento y el socialismo: Reflexiones a partir de la experiencia de la Biotecnología Cubana» Cuba Socialista, 2004, vol., no. 30, pp. 281, ISSN 1852-2300.
2. DELGADO, M., «La calidad y la innovación tecnológica en la biotecnología aplicada a la salud» Dirección y Organización, 1998, vol. 19, no. 19, pp. 125-132, ISSN 1132-175X.
3. SASSON, A. , Biotechnologies in developing countries: present and future, vol. 1, Paris, UNESCO, 1993, ISBN 93 3 102875 8.
4. SPILKER, B., Guide to clinical trials, E.U.A, Raven Press, 1991, ISBN 978-0881677676.
5. «ICH Q10 Pharmaceutical Quality System. Current Step 2 version dated 9 May», [en línea], 2007, [consulta: 2010-09-25], Disponible en: <http://www.emea.eu.int>
6. «ICH Q8 Pharmaceutical Development. Current Step 2 version dated 1 November», [en línea], 2007, [consulta: 2010-09-25], Disponible en: <http://www.emea.eu.int>
7. «ICH Q9 Quality Risk Management. Current Step 4 version dated 9 November», [en línea], 2005, [consulta: 2010-09-25], Disponible en: <http://www.emea.eu.int>
8. DELGADO, M.; VALLÍN, A.; BOLAÑOS, Y.; CORDOVÉS, D. et al., «Gestión Integrada, calidad, medio ambiente y salud del trabajo en la industria biofarmacéutica» Revista Cubana de Gestión Empresarial, 2009, vol. 5, no. 1, ISSN 1682-2455.
9. WECHSLER, J., «Quality: FDA Encourages Quality by Design Initiatives» BioPharm International, March, 2008, vol. 21, no. 3, ISSN 1542-166X.
10. ARORA, T. et al., «Quality:by Design for Biotechnology Products-Part 3" Guidance from the Quality by Design Working Group of the PhARMA Biologics and Biotechnology Leadership Committee on how to apply ICH Q8, Q8R1, Q9 and Q10 to biopharmaceuticals» BioPharm International, 2010, vol. 23, no. 1, ISSN 1542-166X.
11. RATHORE, A.; RON, B.; DOUG, C. , «Quality: Design Space for Biotech Products» BioPharm International, 2007, vol. 20, no. 4, ISSN 1542-166X.
12. STRUCK, M., «Biopharmaceutical. R&D Success Rates and Development Times», Nature Biotechnology [en línea], 1994, vol. 12, pp. 674-677. ISSN (printed): 1087-0156. ISSN (electronic): 1546-1696. Disponible en: <doi:10.1038/nbt0794-674> ;
13. RATHORE, A.; HELEN, W., «Quality by design for biopharmaceuticals. The US Food and Drug Administration's `quality by design' approach is likely to transform the manufacture of biologics», Nature Biotechnology [en línea], 2009, vol., no. 27, pp. 26-34. ISSN (printed): 1087-0156. ISSN (electronic): 1546-1696. Disponible en: <doi: 10.1038/nbt0109-26>;
14. DELGADO, M.; CUEVAS, A., «Guía de la calidad para el registro de vacunas terapéuticas contra el cáncer», Ingeniería Industrial [en línea], 2007, vol. XXVIII, no. 2, ISSN 1815-5936. Disponible en: <http://rii.cujae.edu.cu>
15. HAIR, J.; ROLPH, E.; ANDERSON, R. W. , Análisis Multivariante, 5ta. ed., Madrid, Prentice Hall, 1999, ISBN 84-8322-035-0, p. 832.
Recibido: 11 de enero de 2011
Aprobado: 8 de septiembre de 2011
Daymara González-Fuentes. Centro de Inmunología Molecular (CIM). La Habana, Cuba. Correo electrónico: daymara@cim.sld.cu
