










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Reumatología
versión On-line ISSN 1817-5996
Rev Cuba Reumatol vol.18 no.1 La Habana ene.-abr. 2016
ESTUDIO DE CASOS
Artritis reumatoide y púrpura trombocitopénica asociado a inmunodeficiencia común variable
Rheumatoid arthritis and thrombocytopenic purpura associated with common variable immunodeficiency
Lina Maria Saldarriaga Rivera I, Luis Alberto Delgado Quiroz II, Blanca Elena Ríos Gomes Bica III
I Médica Reumatóloga. Servicio de Reumatología. Hospital Universitario Clementino Fraga Filho, Universidad Federal de Rio de Janeiro, Brasil.
II Médico Reumatólogo. Servicio de Reumatología. Hospital Universitario Clementino Fraga Filho, Universidad Federal de Rio de Janeiro, Brasil.
III Profesora adjunta de la Facultad de Medicina de la Universidad Federal de Rio de Janeiro y jefe de Servicio de Reumatología del Hospital Universitario Clementino Fraga Filho. Universidad Federal de Rio de Janeiro, Brasil.
RESUMEN
La inmunodeficiencia común variable es un trastorno caracterizado por bajos niveles de inmunoglobulinas del suero asociados a una mayor susceptibilidad a infecciones. En algunos pacientes existe una reducción tanto en IgG como en IgA; en otros, los tres principales tipos (IgG, IgA e IgM) de inmunoglobulinas se pueden encontrar disminuidos. Infecciones frecuentes e inusuales pueden ocurrir durante la infancia, pero en la mayoría de los pacientes el diagnóstico es tardío. Enfermedades como artritis reumatoide y púrpura trombocitopénica son trastornos del sistema inmunitario que pueden estar asociados a la inmunodeficiencia común variable.
Palabras clave: artritis reumatoide, púrpura trombocitopénica, inmunodeficiencia común variable, enfermedad autoinmune.
ABSTRACT
Common variable immunodeficiency is a disorder characterized by low levels of serum immunoglobulins associated with increased susceptibility to infections. In some patients there is a reduction in both IgG and IgA, in some others, the reduction is present in three main types of immunoglobulins (IgG, IgA and IgM). Frequent and unusual infections can occur in childhood, but in most patients the diagnosis is delayed. Diseases like rheumatoid arthritis and thrombocytopenic purpura are immune system disorders that can be associated with common variable immunodeficiency.
Keywords: rheumatoid arthritis, thrombocytopenic purpura, common variable immunodeficiency, autoimmune disease.
INTRODUCCIÓN
La inmunodeficiencia común variable (IDCV) engloba un grupo heterogéneo de enfermedades caracterizado por hipogammaglobulinemia de causa desconocida, alteración en la producción de anticuerpos específicos tras inmunización y susceptibilidad a infecciones bacterianas.1
La causa exacta de los niveles bajos de inmunoglobulinas del suero no es conocida. En algunos pacientes existe una reducción tanto en IgG como en IgA; en otros, los tres principales tipos (IgG, IgA e IgM) de inmunoglobulinas se pueden encontrar reducidos.2
Infecciones recurrentes e inusuales pueden ocurrir durante la primera infancia, adolescencia o la vida adulta. En la mayoría de los pacientes, sin embargo, el diagnóstico se realiza en la tercera o cuarta década de la vida.3
Pacientes con IDCV tienen alta predisposición para sufrir enfermedades autoinmunes como tiroiditis de Hashimoto, diabetes, enfermedades del tejido conectivo, púrpura trombocitopénica, enfermedades gastrointestinales y linfoma. La artritis asociada con IDCV puede afectar articulaciones grandes como rodillas, tobillos, codos y muñecas y rara vez afecta las articulaciones pequeñas.4
El diagnóstico diferencial incluye otras causas de hipogammaglobulinemia como pérdida de inmunoglobulinas por vía intestinal o urinaria, e inmunodeficiencia asociada a neoplasias malignas hematológicas, infecciones virales o secundaria a medicamentos.5
El tratamiento de la IDCV se basa en la terapia sustitutiva con inmunoglobulina, administrada por vía intravenosa o subcutánea. Los pacientes con sinusitis crónica o enfermedad pulmonar crónica pueden requerir tratamientos de largo plazo con antibióticos de amplio espectro, mientras que los pacientes con artritis responden favorablemente al tratamiento con inmunoglobulina.6
CASO CLÍNICO
Paciente de 36 años, sexo femenino, raza blanca, natural de Rio de Janeiro, con historia de infección de vías respiratorias altas, otitis, neumonías recurrentes desde los 23 años, que desde el 2007 inicia con cuadro clínico de petequias, especialmente en miembros inferiores, epistaxis, gingivorragia y artralgias, con recuento plaquetario inferior a 150.000 durante los 3 años anteriores, sin ninguna causa hematológica, tóxica o sistémica que la justifiquen, siendo diagnosticada como púrpura trombocitopénica autoinmune. La paciente recibió tratamiento con prednisona 15mg/día, dado que en el momento del diagnóstico se encontraba en estado de gravidez con alto riesgo de sangrado durante el parto.
La paciente continuó en controles en el servicio de hematología, hasta el 2013 cuando inicia cuadro de artritis simétrica bilateral en manos, muñecas, pies y tobillos, rigidez matinal de 1 hora, asociado a fiebre, astenia y adinamia de más de 6 semanas de evolución, por lo que es referida al servició de reumatología.
Al examen físico la paciente presentaba artritis en manos, muñecas, pies y tobillos, bilateral y simétrica, con limitación para la movilidad. Los examenes de laboratorio reportaron Hb:12mg/dl, HCTO:38 %, Leucocitos:5.100/mm3, Plaquetas:109.000/mm3, Neutrófilos: 53 %, linfocitos: 38 %, VHS:14mm/h, PCR:7.3 mg/L (VR:<3mg/L), GOT: 50U/L, GPT:57U/L, GGT:115U/L, Urea:28mg/dl, Creatinina:0.8mg/dl, C3:168mg/dl, C4:31mg/dl, ANA: negativo, Factor Reumatoide: negativo, serología para hepatitis B y C: negativo, Anti-Ro: negativo, Anti-La: negativo, Anti-Sm: negativo, Anti-DNA: negativo, Anti-RNP: negativo, Anti-CCP: negativo. Electroforesis de proteínas: aumento de proteínas alfa-1, alfa-2, beta-1, hipogammaglobulinemia e hipoalbuminemia.
Por la recurrencia de enfermedades infecciosas y presencia de enfermedades autoinmunes se decide investigar el proteinograma electroforético y dosaje de inmunoglobulinas cuyos resultados se muestran en la tabla 1.
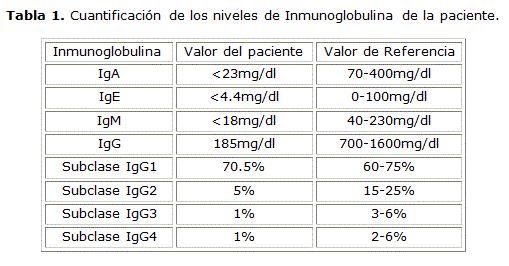
Con estos resultados, se descartó hipótesis diagnósticas como lupus eritematoso sistémico, enfermedad mixta del tejido conectivo, síndrome de sjögren y se diagnosticó la paciente con artritis reumatoide seronegativa e inmunodeficiencia común variable.
La paciente fue tratada con inmunoglobulina intravenosa 6 frascos de 5 g/100ml cada 21 días, prednisona 10mg/día, metotrexato 12.5 mg/semana, ácido fólico 5mg/semana y antibioticoterapia profiláctica para control de las infecciones bacterianas, con remisión de la artritis reumatoide.
DISCUSIÓN
La IDCV es la forma más común de deficiencia severa de anticuerpos que afecta tanto a niños como adultos. La edad de aparición es variable. Por lo general, el diagnóstico se realiza entre las edades de 30 y 40 años de edad. Se estima una prevalencia de 1/25.000 entre los caucásicos afectando por igual a ambos géneros.7
El defecto característico inmune en IDCV se da por alteración en la diferenciación de células B que lleva a una producción defectuosa de inmunoglobulinas. Los niveles séricos de inmunoglobulinas son marcadamente anormales. La IgG en suero suele estar por debajo del límite inferior normal y generalmente por debajo de 400 mg/dL, al igual que los valores de IgA y IgM.8
Un estudio de 248 pacientes con IDCV reportó niveles promedios de inmunoglobulina al momento del diagnóstico: IgG de 258 mg/dl (con un intervalo de referencia normal de 768 a 1728 mg/dL), IgA de 28 mg/dL (rango normal: 99 a 396 mg/dL), IgM de 40 mg/dL (rango normal: 38 a 266 mg/dL).9 Nuestra paciente presentaba niveles más bajos de inmunoglobulinas que lo reportado en la literatura, al igual que una disminución en los niveles de subclase IgG2, IgG3 e IgG4, encontrados en un reporte de caso de la enfermedad indiferenciada del tejido conectivo.10
Alrededor del 25 % de los pacientes con IDCV desarrollan enfermedades autoinmunes, anemia hemolítica, púrpura trombocitopénica idiopática, tiroiditis autoinmune, artritis reumatoide, síndrome de sjögren, artritis idiopática juvenil, artritis reactiva. Estudios demuestran que se desarrolla enfermedad autoinmune hematológica antes o consecutivamente al diagnóstico de IDCV. Otras enfermedades autoinmunes que han sido reportados en pacientes con IDCV incluyen la enfermedad inflamatoria intestinal, hepatitis autoinmune, neutropenia autoinmune y vitíligo.11
En el caso relatado, la alta prevalencia de enfermedades bacterianas en la paciente asociado al cuadro de artritis e hipogammaglobulinemia nos llevó a sospechar en una inmunodeficiencia común variable.
El diagnóstico de artritis reumatoide fue basado en 4 de los 7 criterios propuestos por el Colegio Americano de Reumatología para el diagnóstico de artritis reumatoide (rigidez matinal, artritis de 3 o más regiones articulares, artritis de la articulación de las manos y artritis simétrica). Se obtuvieron 6 puntos en los criterios de clasificación de la artritis reumatoide (ACR/EULAR).
Según la literatura estudiada consideramos que la artritis reumatoide y la púrpura trombocitopénica que presentó la paciente, se encuentran asociadas al síndrome de inmunodeficiencia común variable.
CONCLUSIÓN
La coexistencia de artritis reumatoide, púrpura trombocitopénica e inmunodeficiencia común variable en una paciente, es un caso que hasta el momento tiene pocas descripciones en la literatura.
La reposición de inmunoglobulina combinada con terapia de antibióticos ha mejorado en gran medida la perspectiva de los pacientes con inmunodeficiencia común variable. Los pacientes que presentan solo infecciones bacterianas tienen un mejor pronóstico que aquellos con complicaciones adicionales, como enfermedades autoinmunes lo que representan una causa importante de morbimortalidad.
REFERENCIAS BIBLIOGRÁFICAS
1. Abbott JK, Gelfand EW. Common Variable Immunodeficiency: Diagnosis, Management, and Treatment. Immunol Allergy Clin North Am. 2015;35(4):637-58.
2. Hammarström L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID). Clin Exp Immunol. 2000;120(2):225-31.
3. Saifi M, Wysocki CA. Autoimmune Disease in Primary Immunodeficiency: At the Crossroads of Anti-Infective Immunity and Self-Tolerance. Immunol Allergy Clin North Am. 2015;35(4):731-52.
4. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93(3):190-7.
5. Bonilla FA, Bernstein I, Khan D, Ballas ZK, Chimen J, Frank MM, et al. Practice parameter for diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94:S1-64.
6. Gathmann B, Grimbacher B, BeautéJ, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006-2008. Clin Exp Immunol. 2009;157(Suppl 1):3-11.
7. Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, Koopmans W, Woon ST. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. 2014;15(5):415
8. Seidel MG.Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood. 2014;124(15):2337-44.
9. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34-48.
10. Saldarriaga Rivera LM, Quiroz LAD, Bica BERG. Enfermedad Indiferenciada de Tejido Conectivo asociada a Deficiencia de IgG2 e IgG4. Rev Cubana de Reumatol. 2014;16(01):47-51.
11. Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46(10):1547-54.
Los autores refieren no tener conflicto de intereses
Fuente de financiación: ninguna.
Recibido: 9 de noviembre de 2015
Aprobado: 15 de diciembre de 2015
Publicado: 6 de marzo de 2016
Autor responsable de la correspondencia: Dra. Lina María Saldarriaga Rivera. E-mail: vasculitisreumato@gmail.com
Rua Profesor Rodolpho Paulo Rocco 255. Hospital Universitario Clementino Fraga Filho. Servicio de Reumatología -9º andar. Cidade universitária – Ilha do Fundao. Rio de Janeiro. Brasil. Móvil: 21944-970 Teléfono: +55 21 25622723, +55 21 25622266.

