










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La polimiositis (PM) fue descrita por Wagner en 1882 y en 1887 Ulverrich denominó dermatomiositis a la asociación de dolor y debilidad muscular con lesiones cutáneas inflamatorias. En 1916 Stertz publicó un caso en el que se halló un cáncer gástrico asociado a la dermatomiositis, y señaló la posible relación entre estas entidades.
Arnaud1 señaló que los criterios de Bohan y Peters2 eran útiles para diferenciar las patogenias de estas miopatías, es decir la polimiositis y la dermatomiositis basado en cinco criterios básicos: pérdida de la fuerza simétrica y progresiva de la musculatura proximal de las “cinturas”, con disfagia o no, o afectación de los músculos respiratorios; elevación de las enzimas musculoesqueléticas; biopsia muscular positiva; alteraciones electromiográficas (EMG) y rash cutáneo.
Según el número de criterios presentes podría realizarse un diagnóstico definitivo, probable o posible; tanto la polimiositis y la dermatomiositis deben ser diferenciadas en la actualidad de la miositis por cuerpos de inclusión (MCI), miositis granulomatosas, polimiositis eosinófila, miositis localizada nodular, miositis proliferativa, miositis orbitaria y enfermedades asociadas a miopatías inflamatorias idiopáticas.3 La asociación de la dermatomiositis idiopática y la fibrosis pulmonar está descrita en la literatura. Miller reportó 9 pacientes que presentaron signos sugestivos de fibrosis pulmonar en la radiografía de tórax y en la espirometría elementos de insuficiencia respiratoria restrictiva.4 En la provincia no se habían reportado casos con esta asociación.
Caso clínico
Paciente de 50 años de edad, femenina, mestiza y ama de casa, con antecedentes personales de diabetes mellitus tipo 2 e hipertensión arterial. Lleva tratamiento con insulina NPH 30 unidades diarias, nifedipino 30 mg/día y atenolol 100 mg/día. No hubo antecedentes familiares de interés.
Ingresó porque desde hacía 2 años aquejaba debilidad muscular a nivel de los hombros que le dificulta levantar los brazos por encima de los hombros. Esto le impedía peinarse y realizar labores cotidianas de su casa; además presentaba afectación de la cintura pélvica que le dificultaba levantarse de la posición de sentada y no podía subir escalones. Comentó también debilidad de las rodillas que había favorecido caídas frecuentes, en una de las cuales se fracturó el húmero izquierdo. Igualmente refirió dificultad para deglutir alimentos. Este cuadro clínico se exacerbó, acompañándose de debilidad, anorexia y fatiga.
Exploración clínica
Se halló mucosas húmedas y coloreadas, lesiones pigmentadas de la piel del muslo y la pierna izquierda con atrofia de ambos cuádriceps con predominio del izquierdo (Fig. 1).

Fig. 1- Lesiones pigmentadas de la piel del muslo y la pierna izquierda con atrofia de ambos cuádriceps.
Aparato cardiorrespiratorio: Murmullo vesicular normal, tonos cardiacos rítmicos pero apagados. Tensión arterial 130/80 mmHg. Frecuencia cardiaca central de 88 latidos por min. Abdomen: no se halló visceromegalia. La paciente estuvo orientada en tiempo, espacio y persona, sin signos focales. Manifestó dolores articulares generalizados que involucraban fundamentalmente las grandes articulaciones, con impotencia funcional, y se acompañaban de mialgias generalizadas progresivas, pero más prominentes en la región proximal de ambos brazos, donde llamaba la atención la hipotrofia muscular de varios grupos musculares (Fig. 2).
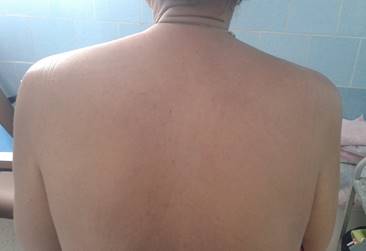
Los reflejos osteotendinosos y la fuerza muscular estaban disminuidas en las cuatro extremidades. El tacto vaginal y rectal fueron normales, y las mamas sin alteraciones. El fondo de ojo resultó normal.
Mediante el estudio analítico se determinó:
Hemoglobina: 120 g/L.
Leucocitos: 10,2 x 109 /L.
Diferencial: normal.
Velocidad de sedimentación globular: 80 mm/h.
Glucemia: 8,2 mmol/L.
Creatinina, lipidograma, ácido úrico y conteo de Addis: normales.
Proteínas totales: albúmina 63,4 %, Alfa1 1,4 %, Alfa 2 2,6 %, Beta 4,7 %, Gamma 12,8 %.
Células LE, factor reumatoideo, VDRL, anticuerpos antinucleares, crioglobulinas, complemento, anticuerpos antifofolípidos: todos negativos.
Enzimas: la creatinfosfokinasa (CPK) se realizó en cuatro oportunidades y osciló entre 800 y 1400 U. El isoenzimograma mostró elevación de la MM. Las transaminasas glutámico oxalacética (TGO) y transaminasas glutámico pirúvica (TGP) realizadas varias veces mostraron un aumento que osciló entre 50 y 155 U. La deshidrogenasa láctica (LDH) en tres ocasiones mostró resultados que oscilaron entre 380 y 500 U, a expensas de las fracciones II y III.
VIH, AgHBs, anticuerpos de hepatitis C, serología lenta en busca de Brucelas y hemocultivos: todos negativos.
Perfil de tiroides: negativo.
Radiografía de tórax posteroanterior: infiltrado difuso peribroncovascular hacia las bases pulmonares asociado a un trayecto fibroso. Mediastino sin alteraciones (Fig. 3).
Radiografía de ambas manos: disminución de espacios articulares interfalángicos bilaterales con cambios degenerativos.
Survey óseo: en el húmero izquierdo se observe una fractura consolidada del tercio distal de la diáfisis humeral, el resto del examen fue normal.
Ultrasonido abdominal y ginecológico: sin alteraciones.
Electrocardiograma: normal.

Tomografía axial computadorizada de tórax y abdomen: No alteraciones mediastinales. Se halló focos de infiltración intersticial bilateral en los tercios inferiores con discreto engrosamiento pleural hacia la periferia (Fig. 4). El resto de las estructuras abdominales y ginecológicas sin alteraciones.

Espirometría: trastornos ventilatorios restrictivos de grave intensidad.
Electroneurofisiología (ENF) del cubital, mediano y tibial posterior: normales.
Electromiografía tibial anterior derecho e interóseos derechos: presentan fibrilación espontánea que aparece en el reposo.
Potencial de unidad motora (PUM): mostró pequeña amplitud y corta duración.
Electroencefalograma: normal.
Biopsia de piel: infiltrado perivascular de linfocitos del plexo profundo.
Biopsia de músculo deltoides: mostró un patrón de afectación inflamatorio (Fig. 5).

Fig. 5- Infiltrado perivascular de linfocitos del plexo profundo. Obsérvese la presencia de linfocitos en la periferia de los cuatro fascículos y alrededor de los vasos con atrofia de fibras musculares de predominio perifascicular.
La paciente fue tratada con prednisona a 1 mg/kg/día como dosis inicial, y se redujo a 5 mg semanales, asociado con azatioprina a dosis de 1,5 mg/kg/día. Este tratamiento fue muy eficaz, ya que la paciente presentó una notable recuperación, tanto clínica como por estudios de laboratorio. La paciente se negó a realizarse endoscopía de esófago, estómago y duodeno, así como colonoscopía.
Discusión
El diagnóstico de una miopatía inflamatoria debe considerase cuando los pacientes presentan debilidad de las extremidades superiores e inferiores, sin síntomas sensoriales o con las lesiones típicas características de la dermatomiositis,1,4 menos frecuentemente la elevación de las cifras de la CPK, lo que guia al diagnóstico de miopatía inflamatoria.5
Los pacientes con dermatomiositis presentan lesiones de piel o músculos características, con gran debilidad de estas últimas.4,5,6
Las lesiones patognomónicas en la piel son el rash en “heliotropo”, que es un rash violáceo periorbitario con edema macular. En otras ocasiones se presenta el signo de Gottron, constituido por pápulas violáceas que se presentan en el dorso de las articulaciones metacarpofalángicas e interfalángicas de las articulaciones de ambas manos. Igualmente puede haber telangiectasias periungueales, trombosis de capilares, poiquilodermia y áreas de fotosensibilidad,6 en zonas de exposición en la parte superior de la espalda conocido como el “signo del mantón”. Aparece un prurito intenso y molesto, al igual que diferentes tipos de eritemas.1,4,5 Estos dos últimos fueron los encontrados en esta paciente. La debilidad muscular es muy frecuente pero inespecífica, pues se presenta en diferentes tipos de enfermedades de los músculos.7
Al inicio el cuadro clínico simula una miopatía en el curso de una enfermedad autoinmune, ya que este caso debutó de la forma clásica de la dermatomiositis con lesiones dérmicas que preceden el desarrollo de la miopatía.3,4 Algunos enfermos tienen una dermatomiositis amiopática con cifras normales de enzimas musculares, resonancia magnética e incluso biopsia de músculo negativa.8
La miopatía afecta los músculos proximales, es simétrica y se acompaña de fatiga, debilidad, mialgias y dolores articulares.1,3,4,7 Como observación en esta paciente existían otros síntomas y signos que pueden presentarse como neumonitis intersticial, afección cardiaca, vasculitis, disfagia, artralgia y artritis.7 Observamos los trastornos respiratorios, disfagia, artralgia y artritis.
Se señala que las alteraciones enzimáticas propias de la dermatomiositis plantean problemas diferenciales con los observados en enfermedades malignas, hepatopatías e infarto del miocardio, por lo que se considera que la asociación de la elevación de la TGP, CPK y de la fracción II y III de la LDH afirman el diagnóstico de dermatomiositis,4 como pudimos apreciar en el estudio de esta enferma.4,7
Las alteraciones de la EMG que suelen observarse en la dermatomiositis-polimiositis son:9,10,11,12
Fibrilación espontánea de aparición en el reposo.
Potencial de unidad motora (PUM) de pequeña amplitud y corta duración.
Salvas de potenciales repetitivos “seudomiotónicos”, de alta frecuencia por estimulación mecánica.
La EMG de esta enferma fue concordante con el presunto diagnóstico. La dermatomiositis-polimiositis en asociación con procesos malignos son síndromes paraneoplásicos, pero su tratamiento, pronóstico y factores predisponentes no están aún claros.
Algunos autores8 señalan la incidencia de polimiositis-dermatomiositis y neoplasias entre un 13-15 % y otros6 reportan entre el 20-30 %. Los datos ofrecidos por la biopsia de piel y músculo muestran el patrón clásico de afectación dermatomiositis-polimiositis y puede ocurrir atrofia perifascicular. lo que ofrecen el diagnóstico como observamos en nuestra paciente.
Los esteroides son el tratamiento de elección.1,3,4 La prednisona a dosis de 1 mg/kg/día en dosis inicial de 60 mg es lo más aconsejable, transcurridos 6-8 semanas de tratamiento puede iniciarse una reducción lenta de la dosis. El tratamiento debe durar de 12-18 meses.3,4,9 En los casos muy graves pueden asociarse agentes inmunosupresores como la ciclofosfamida, azatioprina, metrotrexate y cloranbucilo.3,4,11,12 En este caso usamos esteroides y azatioprina, esta última a dosis de 1,5 mg/kg/día, con lo que se logró una gran eficacia por la notable mejoría clínica y de los niveles de enzimas séricas.
Conclusiones
Los criterios diagnósticos en la dermatomiositis están bien establecidos, fundamentalmente los clínicos. Si bien la fibrosis pulmonar es una manifestación extraarticular de la enfermedad, es imprescindible realizar el diagnóstico de esta complicación para mejorar el pronóstico del paciente y su calidad de vida. Sin embargo, en la práctica médica los hallazgos dermatológicos y los estudios histopatológicos de la biopsia de músculo dan como resultado el diagnóstico; además los exámenes de la función respiratoria y los estudios imagenológicos confirman la presencia de esta complicación.

