










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkCorSalud
versión On-line ISSN 2078-7170
CorSalud vol.9 no.1 Santa Clara ene.-mar. 2017
Amiloidosis cardíaca. A propósito de un caso
Cardiac amyloidosis. Apropos of a case
Wendy Fusté Pedroso1, Carlos Ramos Emperador1, Mariam González Gorrín1, Reinaldo Milán Castillo2, Daisy Luperón Loforte2, Ángela Castro Arca2, Juan A. Prohías Martínez2, Wanda Fusté Pedroso3
1. Servicio de Cardiología. Hospital Clínico-Quirúrgico Hermanos Ameijeiras. La Habana, Cuba. Correo electrónico: wendyfoste@infomed.sld.cu
2. Departamento de Ecocardiografía. Hospital Clínico-Quirúrgico Hermanos Ameijeiras. La Habana, Cuba.
3. Centro de Neurociencias de Cuba. La Habana, Cuba.
RESUMEN
La amiloidosis cardíaca es una enfermedad grave producida por el depósito extracelular de amiloide a ese nivel. Su diagnóstico y tratamiento depende de la identificación histopatológica de los depósitos de amiloide y de la determinación inmunohistoquímica, bioquímica o genética de su tipo. Los antecedentes del paciente, su edad, la presentación clínica, la exploración física, el aparato o sistema afectado, las causas subyacentes y los antecedentes familiares pueden proporcionar indicios útiles sobre el tipo de amiloide. En este trabajo se hace referencia a un caso de amiloidosis cardíaca en un anciano cuya presentación clínica inicial estuvo marcada por episodios sincopales. Ante la dificultad para llegar al diagnóstico del subtipo de amiloidosis con el empleo de estudios de alta tecnología, se refleja la utilidad de los datos clínicos, como elemento decisivo que nos pueden orientar a sospechar el diagnóstico.
Palabras clave: amiloidosis, corazón, arritmias cardíacas, síncope.
ABSTRACT
Cardiac amyloidosis is a serious disease caused by extracellular amyloid deposition at that level. Its diagnosis and treatment depends on histopathologically identifying amyloid deposits, and immunohistochemically, biochemically or genetically determining its type. Patient's history, age, clinical presentation, physical examination, affected organ system or apparatus, underlying causes and family history, may provide useful clues to the amyloid type. In this paper we discuss a case of cardiac amyloidosis in an elderly patient whose initial clinical presentation was marked by syncopal episodes. Difficulties in diagnosing amyloidosis subtype by using high technology studies indicate the usefulness of clinical data as a crucial element that may lead us to suspect diagnosis.
Key words: amyloidosis, heart, cardiac arrhythmias, syncope.
INTRODUCCIÓN
La amiloidosis es el término para un grupo de trastornos en el plegamiento de las proteínas que se caracteriza por el depósito de fibrillas insolubles de polímeros proteínicos en tejidos y órganos1.
El término amiloide fue acuñado en 1854 por el patólogo Rudolf Virchow, quien consideraba que estos depósitos, a la observación en el microscopio, tenían un aspecto similar al almidón (del latín amylium)1. En la amiloidosis, los agregados por lo general, son extracelulares y las unidades proteínicas mal plegadas asumen una conformación estructural antiparalela, con plegamiento â en forma de hojas, lo que lleva a la formación de oligómeros de orden elevado y a la consiguiente formación de fibrillas con propiedades singulares de tinción. Se ha identificado su policromatismo con cristal violeta, el aspecto violáceo con el ácido periódico de Schiff, aunque el criterio histológico más utilizado es la positividad para el rojo Congo (donde se observa una birrefringencia verde-manzana característica en el estudio de luz polarizada)1,2.
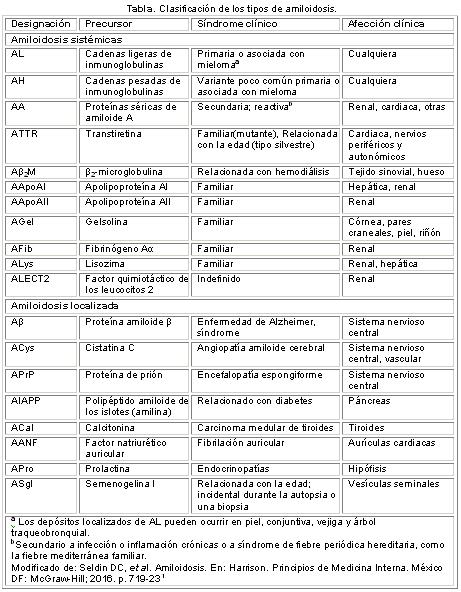
Se conocen disímiles proteínas que pueden producir depósitos amiloides en distintos tejidos y dan lugar a diversas afecciones que varían desde la fibrosis quística hasta la enfermedad de Alzheimer (Tabla). De estas solo algunas producen afección cardíaca1-4.
CASO CLÍNICO
Hombre de 76 años de edad, con antecedentes de hipertensión arterial (HTA) de 17años de evolución, operado por hiperplasia prostática benigna, que había sido hospitalizado un mes antes del ingreso por un accidente vascular encefálico que no dejó secuelas neurológicas. Fue remitido para valoración cardiológica tras haber presentado 2 episodios de síncope en las últimas 3 semanas. Estos episodios se acompañaron de relajación de esfínteres y respiración agónica, con recuperación espontánea en pocos minutos. No se recogieron antecedentes familiares de cardiopatía o muerte súbita.
En la anamnesis el paciente refirió disnea de esfuerzo, clase funcional II de la New York Heart Association (NYHA) de meses de evolución, y estabilidad clínica, sin ortopnea, ni episodios de disnea paroxística nocturna. El día antes al ingreso comenzó a presentar tos pertinaz nocturna. A su llegada al centro médico, en la exploración física llama la atención la presencia de ingurgitación yugular, latido de la punta desplazado hacia la izquierda, soplo sistólico en foco mitral II/VI y crepitantes en bases pulmonares, con cifras de tensión arterial de 100/60 mmHg. Se le realizó electrocardiograma que evidenció fibrilación auricular con frecuencia ventricular adecuada y bloqueo fascicular anterior izquierdo.
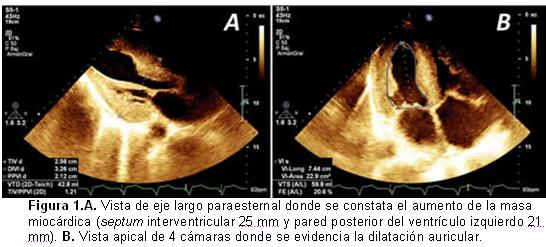
El ecocardiograma transtorácico mostró un engrosamiento de las paredes ventriculares (Figura 1A), con patrón granular del miocardio y disfunción diastólica tipo III. La fracción de eyección del ventrículo izquierdo, evaluada por el método de Simpson, fue de 20% y se constataron, además, dilatación biauricular a predominio de la derecha (Figura 1B), hipertensión pulmonar moderada, derrame pericárdico laminar e insuficiencia mitral moderada.
Se realizó un Holter de 24 horas, que demostró fibrilación auricular con respuesta ventricular entre 100-105 latidos por minuto, complejos ventriculares prematuros aislados, a veces bigeminados, con 4 episodios de taquicardia ventricular no sostenida.
La coronariografía confirmó la existencia de arterias coronarias sin lesiones angiográficas y se realizó un cateterismo derecho con toma de muestra para biopsia endomiocárdica, donde se evidenció la infiltración miocárdica difusa por depósito de amiloide (Rojo Congo positivo), sin ser posible confirmar el subtipo mediante las técnicas de inmunohistoquímica empleadas.
Se completó el estudio con un análisis de sangre cuyos valores estuvieron dentro de los límites normales, a excepción de la enzima deshidrogenasa láctica (LDH) que estaba levemente elevada, lo cual se asocia a la disfunción cardíaca por la insuficiencia aguda y las arritmias asociadas.
La biopsia de médula ósea fue normal. Las cadenas ligeras kappa y lambda, la inmunofijación en suero y orina no fueron evaluadas por carencia de reactivos.
La radiografía de tórax evidenció leve derrame pleural bilateral, con predominio derecho. El ultrasonido abdominal mostró leve hepatomegalia global, homogénea, con leve dilatación de las venas suprahepáticas y la cava inferior, sugestivo de congestión. Vesícula de paredes finas con múltiples litiasis y riñón derecho con dos formaciones quísticas.
Se definió el diagnóstico de miocardiopatía infiltrativa amiloidótica y se decidió la implantación de un cardiodesfibrilador automático (CDAI) unicameral, procedimiento que se realizó sin complicaciones, y se mantuvo el tratamiento con diuréticos (furosemida y espironolactona), digoxina y anticoagulación oral (warfarina). El paciente evolucionó favorablemente y fue egresado con seguimiento ambulatorio por consulta externa.
COMENTARIO
No fue posible determinar el origen de la amiloidosis por limitaciones de recursos, aunque existe una alta sospecha clínica de que se tratase de amiloidosis cardíaca senil por implicación de la proteína precursora transtiretina (TTR). La confirmación requiere de estudios como una gammagrafía con tecnecio difosfato (Tc-DPD), lo que mostraría una intensa captación difusa del radio trazador a nivel biventricular, la secuenciación del gen de transtiretina sin alteraciones genéticas y la biopsia endomiocárdica, que confirmaría por inmunohistoquímica y por espectrometría de masas que el precursor amiloide implicado era la TTR3.
Este caso clínico refleja la dificultad que se presenta al definir el subtipo de amiloidosis cardíaca. Aunque el diagnóstico de certeza se establece con el análisis que se ha mencionado de la biopsia endomiocárdica, así como por estudios genéticos; estas técnicas solo están disponibles en centros expertos y habitualmente su interpretación es difícil. Por tales razones una vez más adquiere valor la clínica para guiar la sospecha del subtipo de amiloidosis y es imprescindible dominar con precisión las diferentes variantes que pueden provocar afección cardíaca significativa.
Aunque varios tipos de amiloide pueden infiltrar el corazón, solo la variedad primaria (AL, Tabla), la secundaria (AA), la senil, y algunas formas de las hereditarias, pueden producir manifestaciones clínicas cardiovasculares significativas5,6.
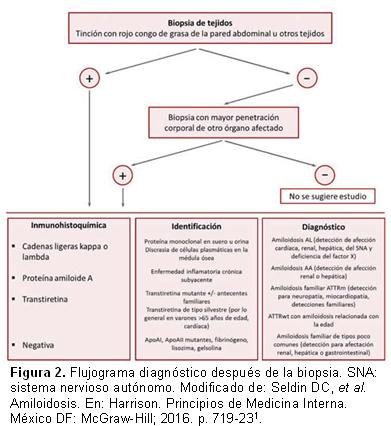
Cada subtipo tiene una evolución, tratamiento y pronóstico diferentes. Por este motivo, ante un paciente que padece amiloidosis cardíaca es muy importante establecer correctamente de qué subtipo se trata (Figura 2).
En el anexo se describe una sucinta revisión de algunos tipos de amiloidosis que pueden afectar al corazón.
Manifestaciones cardíacas
Las manifestaciones clínicas cardíacas son similares independientemente del subtipo de amiloidosis. Es frecuente el desarrollo de insuficiencia cardíaca (IC) diastólica con predominio de signos congestivos y, aunque las presiones en el ventrículo izquierdo están elevadas, el edema de pulmón es infrecuente.
Algunos pacientes sufren angina en relación con la infiltración amiloide vascular. En ocasiones la angina se acompaña también de claudicación mandibular. Los vasos afectados son normalmente intramiocárdicos y las arterias epicárdicas no suelen mostrar lesiones7.
La presencia de síncope o presíncope es habitual, como resultado de la combinación de disfunción autonómica y arritmias, en un corazón con poca reserva funcional. Los depósitos alteran la regulación adrenérgica del corazón, así como la regulación basal y la respuesta cardíaca a la estimulación neurohormonal. Frecuentemente se observa desaparición de la HTA si el paciente era previamente hipertenso, con desarrollo de hipotensión postural secundaria a la excesiva diuresis o la neuropatía autonómica1,8.
La aparición de síncope de esfuerzo tiene un pronóstico infausto, ya que es un marcador de miocardiopatía restrictiva grave y se asocia a una mortalidad elevada en los 3 meses siguientes, normalmente en relación con muerte súbita9,10.
La fibrilación auricular es frecuente debido a la dilatación progresiva de las aurículas y la fisiología restrictiva, tal como ocurre en este paciente; además, se asocia a una alta incidencia de fenómenos tromboembólicos6.
Aunque las arritmias ventriculares son frecuentes, son causa infrecuente de síncope y no suelen ser el síntoma de presentación, aunque en este caso fue la manifestación clínica por la que el paciente acudió en busca de atención médica: síncope asociado a arritmia ventricular (taquicardias ventriculares no sostenidas).
Los bloqueos aurículo-ventriculares de alto grado y la disfunción sintomática del nodo sinusal son raros. La muerte súbita, aunque frecuente en estos pacientes, suele deberse más a disociación electromecánica que al desarrollo de arritmias ventriculares11,12.
Hallazgos complementarios
Electrocardiograma: Es característico observar una disminución del voltaje, a pesar del aumento del grosor de la pared miocárdica, y aparecerá el tipo de arritmia si existiera. Las más frecuentes son: fibrilación auricular, bloqueos aurículo-ventriculares y taquicardia ventricular12,13.
Ecocardiografía: Se pueden encontrar diferentes signos de la enfermedad, aunque los clásicos sólo son propios de sus fases avanzadas. El hallazgo más precoz es el engrosamiento de la pared ventricular izquierda (particularmente en ausencia de HTA), con evidencia de disfunción diastólica asociada, que es el hallazgo ecocardiográfico por antonomasia de esta enfermedad y está presente, en algún grado, en prácticamente todos los pacientes. El miocardio presenta una textura granular, dato que sólo es valorable en ausencia de segundo harmónico. Los hallazgos característicos de la amiloidosis cardíaca avanzada son hipertrofia biventricular, engrosamiento de las válvulas y del tabique interauricular, dilatación auricular, derrame pericárdico pequeño y disfunción sistólica. El ecocardiograma Doppler muestra un patrón de llenado restrictivo del ventrículo izquierdo1,6,12. El uso de Doppler tisular y la deformación miocárdica en el tiempo (strain rate) pueden ser de especial interés en esta enfermedad. Así, mediante el primero se encuentra reducción en las velocidades diastólicas en fases tempranas y tardías de la enfermedad, y permite su identificación incluso cuando prácticamente no hay hipertrofia ventricular. El segundo, permite documentar precozmente la afección cardíaca al detectar un deterioro en la función contráctil longitudinal, y sus hallazgos se han relacionado con el desarrollo posterior de IC y con la posibilidad de diferenciar distintos tipos de amiloidosis12.
Resonancia magnética nuclear: Existen varios patrones de realce tardío con gadolinio indicativos de esta enfermedad, pero es el realce subendocárdico global en anillo el que se presenta con más frecuencia y el que se ha asociado a amiloidosis cardíaca14. Un patrón de realce tardío extenso y transmural orienta hacia la amiloidosis por TTR15. Las pruebas de medicina nuclear con el componente P del amiloide sérico tienen la limitante de no detectar el depósito amiloideo en el tejido cardíaco9. La gammagrafía con Tc-DPD es útil para diferenciar amiloidosis TTR (formas hereditaria o senil) de la amiloidosis AL, y otras miocardiopatías que cursan con hipertrofia ventricular izquierda. Los corazones de los pacientes con amiloidosis TTR captan 99mTc-DPD, muestran un intenso depósito cardíaco de forma biventricular, mientras que aquellos con amiloidosis AL no lo suelen captar15. Con la comercialización de nuevos marcadores específicos de los depósitos de amiloide (como el 18F-florbetapir), se pueden obtener imágenes de las cadenas ligeras de amiloide en el miocardio16.
Tratamiento
El trasplante hepático ortotópico reduce la progresión de la enfermedad, aunque no corrige la neuropatía sensitivo-motora y es más exitoso en pacientes jóvenes con neuropatía periférica temprana. La TTR puede estabilizarse por la unión a fármacos como el diflunisal, el ácido flumenámico,o el tafamidis, sustancias que están siendo estudiadas actualmente y en el caso del diflunisal se plantea que reduce la progresión de la polineuropatía1,17. En la amiloidosis senil el tratamiento es sintomático9.
Amiloidosis primaria
Se asocia a la discrasia de las células plasmáticas como mieloma, linfoma. El amiloide es generado a partir de un clon celular que produce de forma anormal cadenas ligeras (LC) de inmunoglobulinas. Generalmente ocurre después de los 40 años y, a menudo, es rápidamente progresiva y letal sin tratamiento.
Los depósitos de amiloide pueden presentarse en el intersticio de cualquier órgano fuera del sistema nervioso central. Los afectados en la clínica pueden someterse a biopsia. Por lo general, se examinan los vasos sanguíneos de las encías o de la mucosa rectal, pero el tejido accesible con mayor facilidad (positivo en >80% de los pacientes) es la grasa. Si el material es negativo, puede considerarse un procedimiento con mayor penetración corporal con biopsias de riñón, corazón, hígado o del tubo digestivo1,6.
Cursa con un cuadro florido que abarca desde síntomas inespecíficos como fatiga y pérdida de peso, hasta muy específicos en dependencia del órgano afectado. La afección cardíaca es la principal causa de muerte.
La macroglosia es un signo patognomónico, pero se observa sólo en un 10% de los pacientes. La afección hepática causa colestasis y hepatomegalia. El bazo a menudo está afectado y puede haber hipoesplenismo funcional con ausencia de esplenomegalia significativa. Otros datos incluyen: equimosis cutáneas (particularmente periorbitarias o signo de «ojos del mapache»), distrofia ungueal, alopecia y artropatía con engrosamiento de las membranas sinoviales en las muñecas y los hombros1,6.
La mediana de supervivencia sin tratamiento suele ser sólo de uno a dos años a partir del momento del diagnóstico, y en casos de afección amiloide del miocardio de unos ocho meses1,6,11.
El tratamiento se basa en la quimioterapia y el trasplante de la médula ósea.
Los diuréticos y el uso de medias elásticas pueden disminuir el edema, los inhibidores de la enzima convertidora de angiotensina deben utilizarse con precaución y no han demostrado reducir la progresión de la enfermedad renal. Puede facilitarse la diuresis eficaz con la administración de albúmina para incrementar la presión oncóticaintravascular1,12.
La IC congestiva se trata mejor con diuréticos. Los digitálicos, anticálcicos y los betabloqueadores presentan contraindicaciones relativas pues pueden interactuar con las fibrillas de amiloide y producir bloqueo cardíaco y deterioro de la IC. Se ha utilizado amiodarona para las arritmias auriculares y ventriculares. Los desfibriladores automáticos implantables tienen menor eficacia por el engrosamiento del miocardio, pero pueden ser beneficiosos para algunos pacientes. Para las anomalías de la conducción puede indicarse el uso de marcapasos. La disfunción contráctil de las aurículas es común y es indicación para anticoagulación, incluso en ausencia de fibrilación auricular1,7,11.
Amiloidosis reactiva
El amiloide AA está compuesto por proteínas séricas de amiloide A (SAA) reactivas de fase aguda, lo cual ocurre en el caso de enfermedades inflamatorias crónicas o infecciosas.
La afectación de los órganos por lo general inicia en los riñones. También pueden ocurrir hepatomegalia, esplenomegalia y neuropatía del sistema nervioso autónomo conforme progresa la enfermedad; también ocurre miocardiopatía, aunque con poca frecuencia1,6,7.
Amiloidosis familiar y senil
La amiloidosis familiar se debe, a menudo, a mutaciones en la proteína precursora TTR. Esta molécula, con síntesis principalmente hepática, circula por la sangre y el líquido cefalorraquídeo actuando como transportador de la tiroxina (T4) y de la proteína de unión al retinol6.
Es una enfermedad genética, con herencia autosómica dominante y elevada penetrancia. El órgano afectado de forma predominante va a depender del tipo de mutación, y existen 14 formas que producen afectación cardíaca. Otras amiloidosis familiares, causadas por variantes de apolipoproteínas, las AI o AII, gelsolina, fibrinógeno Aá con lisozima, se han informado sólo en unas cuantas familias en todo el mundo1,6,8.
Se presenta como un síndrome de polineuropatía con miocardiopatía amiloidótica familiar. La neuropatía periférica inicia en fibras sensitivas pequeñas de las extremidades inferiores, y neuropatía motora que progresa hacia las extremidades superiores. La neuropatía autonómica se manifiesta con diarrea, pérdida de peso e hipotensión ortostática. Las opacidades del vítreo causadas por depósitos de amiloide son patognomónicas de esta variedad18,19.
Sin intervención, el período de supervivencia después de su inicio es de 5 a 15 años. La secuenciación de DNA es el método estándar para el diagnóstico genético1.
Los depósitos amiloides sin significación clínica a nivel cardíaco son muy frecuentes en los ancianos. Pero en ocasiones estos depósitos son masivos y generan un compromiso de la función cardíaca con repercusión clínica, que da lugar a la amiloidosis senil, debida al depósito de TTR no mutada6. Esta enfermedad afecta casi exclusivamente a varones de edad avanzada, y es excepcional por debajo de los 60 años. A diferencia de otras formas de amiloidosis, la afección de otros órganos es rara (exceptuando la presencia de síndrome del túnel carpiano y estenosis del canal lumbar). La clínica habitual es la aparición de IC y cardiomegalia9,10,18,19.
Pese a la avanzada edad de los pacientes y la gran infiltración cardíaca, la IC es de más fácil control y su mediana de supervivencia es de 75 meses, muy superior a la de otros tipos de amiloidosis. En cualquier caso, la muerte de estos pacientes suele estar en relación con la progresión de la IC y la aparición de arritmias4.
CONFLICTOS DE INTERESES
Los autores declaran que no existen conflictos de intereses.
REFERENCIAS BIBLIOGRÁFICAS
1. Seldin DC, Berk JL. Amiloidosis. En: Kasper DL, Hauser SL, Jameson JL, Fauci AS, Longo DL, Loscalzo J, eds.Harrison. Principios de Medicina Interna, 19ª ed. México DF: McGraw-Hill; 2016. p.719-23.
2. González I, Jadue N, Trejo C, Carvallo A. Amiloidosis: Revisión a propósito de un caso clínico. Rev Chil Reumatol. 2010;26:285-9.
3. Fikrle M, Palecek T, Kuchynka P, Nemecek E, Bauerova L, Straub J, et al. Cardiac amyloidosis: A comprehensive review. Cor et Vasa. 2013;55:e60-75.
4. Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203-12.
5. Calero Núnez S, Tercero Martínez A, García López JC, Jiménez-Mazuecos J. Amiloidosis cardiaca senil y estenosis aórtica degenerativa: 2 enfermedades interrelacionadas en el anciano. Rev Esp Geriatr Gerontol. 2017;52:167-70.
6. Barge-Caballero G, Couto-Mallón D, Barge-Caballero E, Paniagua-Martín MJ, Barriales-Villa R, Pombo-Otero J, et al. ¿Cómo enfrentarse a una sospecha clínica de amiloidosis cardíaca? Un enfoque práctico para el diagnóstico. Cardiocore. 2017;52:27-34.
7. Gómez-Bueno M, Segovia J, García-Pavía P, Barceló JM, Krsnik I, Sánchez-Turrión V, et al. Amiloidosis cardiaca: la importancia del manejo multidisciplinario. Rev Esp Cardiol. 2009;62:698-702.
8. Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol.2010;7:398-408.
9. Idiazabal U, García-Pavía P, Azcárate PM, Idoate F, Mercado MR. Amiloidosis cardiaca por transtiretina: la gammagrafía mostró el camino. Rev Colomb Cardiol. 2016;23:71.e1-5.
10. Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40:232-9.
11. Vargas Vergara D, Olaya A, Andrade RE, Mora G. Bloqueo auriculoventricular completo como manifestación de amiloidosis cardiaca. Rev Colomb Cardiol. 2016;23:150.e1-5.
12. García-Pavía P, Tomé-Esteban MT, Rapezzi C. Amiloidosis. También una enfermedad del corazón. Rev Esp Cardiol. 2011;64:797-808.
13. Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol. 2014;114:1089-93.
14. Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3:155-64.
15. Dungu JN, Valencia O, Pinney JH, Gibbs SD, Rowczenio D, Gilbertson JA, et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7:133-42.
16. López-Fernández T, Saura D, Rodríguez-Palomares JF, Aguadé-Bruix S, Pérez de Isla L, Barba-Cosials J. Selección de temas de actualidad en imagen cardiaca 2015. Rev Esp Cardiol. 2016;69:286-93.
17. Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22:79-83.
18. Westermark P, Westermark GT, Suhr OB, Berg S. Transthyretin-derived amyloidosis: Probably a common cause of lumbar spinal stenosis. Ups J Med Sci. 2014;119:223-8.
19. Yanagisawa A, Ueda M, Sueyoshi T, Okada T, Fujimoto T, Ogi Y, et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol. 2015;28:201-7.
Recibido: 21 de enero de 2017
Aceptado: 23 de febrero de 2017
Wendy Fusté Pedroso. Servicio de Cardiología. Hospital Clínico-Quirúrgico Hermanos Ameijeiras. La Habana, Cuba. Correo electrónico: wendyfoste@infomed.sld.cu

