










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El término diabetes mellitus (DM) describe un desorden metabólico de múltiples etiologías, caracterizado por hiperglucemia crónica con trastornos en el metabolismo de los carbohidratos, grasas y proteínas a causa de los defectos en la secreción y/o en la acción de la insulina o de ambos. 1,2
La insulina es una hormona peptídica de 5.8 KDa y es secretada por las células beta en los islotes pancreáticos de Langerhans en respuesta a niveles elevados de nutrientes en la sangre. Su principal función es la de mantener la concentración de glucosa en sangre en un rango normal. En el tejido hepático inhibe su producción y favorece el almacenamiento de esa molécula. Además, regula el metabolismo de los carbohidratos, lípidos y proteínas y promueve la división y el crecimiento celular a través de sus efectos mitogénicos. 3 El apropiado almacenamiento y liberación de energía durante los estados de alimentación y ayuno son esenciales para la sobrevivencia y son controlados principalmente por la acción de esta hormona. 4) Las acciones de la insulina son mediadas por cascadas de señalización intracelular, las cuales pueden sufrir modificaciones que alteran el normal funcionamiento del organismo. La hiperglucemia concomitante puede determinar una serie de cambios irreversibles en la composición química de las moléculas, como la glicosilación no enzimática que favorece el estrés oxidativo. Estas reacciones ocurren de forma acelerada en la DM, avalando los cambios morfofuncionales que se producen en los individuos diabéticos como las complicaciones microvasculares y macrovasculares que aparecen en esta entidad. 3
El conocimiento de la relación entre las bases moleculares de la DM con la glicosilación no enzimática y el estrés oxidativo conduce a una mejor interpretación de esta entidad como la resistencia a la insulina y sus complicaciones crónicas, además de otras relacionadas como el envejecimiento, el cáncer y desarrollo de enfermedades degenerativas como el Alzheimer. Todo ello repercute en una mejor atención a los pacientes sobre todo en los aspectos relacionados con el diagnóstico y tratamiento de estas enfermedades.
Desarrollo
Mecanismos moleculares de las acciones de la insulina
La insulina es un péptido de 51 aminoácidos producido y secretado por las células beta de los islotes pancreáticos. Está constituida por dos cadenas polipeptídicas, A y B, de 21 y 30 aminoácidos, respectivamente, conectadas por puentes disulfuro. Sus acciones biológicas se inician cuando se une con su receptor, una glucoproteína integral de membrana, la cual está formada por dos subunidades alfa y dos subunidades beta. La subunidad alfa, de 135 kDa, que contiene el sitio de unión para la insulina, es totalmente extracelular y se une a la región extracelular de la subunidad beta, así como a la otra subunidad alfa, a través de puentes disulfuro. La subunidad beta, de 95 KDa, se compone de un dominio extracelular, uno transmembranal y uno intracelular de cinasa, que es activado por autofosforilación. El receptor de insulina pertenece a la familia de receptores con actividad intrínseca de cinasa de tirosinas (Tyr). La unión de la insulina a la subunidad alfa del receptor genera cambios conformacionales que inducen su activación catalítica y a la autofosforilación de varios residuos de Tyr localizados en la región citosólica de la subunidad beta. Los residuos autofosforilados son entonces reconocidos por diferentes proteínas adaptadoras, entre las que se incluyen miembros de la familia del sustrato del receptor de insulina (IRS), de los cuales el IRS-1 y el IRS-2 constituyen los dos principales sustratos e intermediarios más comunes en la etapa inicial de propagación de la señal de insulina. El IRS actúa como una molécula adaptadora que organiza la formación de complejos moleculares y desencadena cascadas de señalización intracelular. La mayoría de las acciones de la insulina se llevan a cabo mediante la activación de dos vías principales de señalización: la vía de la fosfatidilinositol-3-cinasa (PI3K)/Akt también llamada proteína cinasa B (PKB), responsable de la mayoría de sus acciones metabólicas y la vía de las cinasas activadas por mitógeno/Ras (MAPK/Ras), que regula la expresión genética y los efectos mitogénicos asociados a la insulina. 3
En el caso de la vía PI3K/Akt, la cinasa Akt desempeña un papel central en la señalización de la insulina, ya que su activación lleva a la fosforilación de un número de sustratos con funciones clave en una amplia variedad de procesos biológicos, entre los que se incluyen enzimas, factores de transcripción, proteínas reguladoras del ciclo celular y proteínas de apoptosis y sobrevivencia.
Se han identificado tres isoformas de Akt (Akt1, 2 y 3), de las cuales Akt2 parece desempeñar un papel importante en las acciones metabólicas de la insulina, incluyendo la incorporación de glucosa en el músculo y el tejido adiposo a través de la translocación de GLUT-4 de compartimentos intracelulares a la membrana celular, para aumentar la captación de glucosa en la célula. 3
Además, Akt participa en la síntesis de glucógeno a través de la inhibición de GSK-3β, de proteínas vía blanco de rapamicina en mamíferos/cinasa de la proteína ribosomal S6 de 70 kDa y de lípidos. Ras-GTP opera como un interruptor molecular, estimulando la activación de la cascada de MAPK, a través de la activación secuencial de Raf, MEK y ERK1/2. Una vez activas, ERK1/2 se translocan al núcleo y catalizan la fosforilación de factores de transcripción que regulan la expresión génica y promueven el crecimiento, la proliferación y la diferenciación celular. (3
Regulación de la señal de la insulina
Las acciones metabólicas y promotoras del crecimiento y proliferación celular de la insulina son reguladas de manera precisa a través de mecanismos de autorregulación (regulación homóloga), en donde las enzimas activadas por la propia vía, inhiben la actividad de proteínas claves de la señalización de la insulina. De forma adicional, existen mecanismos moleculares homeostáticos, no relacionados con los activados por la insulina, que pueden inhibir también la señalización de esta hormona (regulación heteróloga). Ambos mecanismos son de suma importancia, ya que mantienen el estado de homeostasis celular, definiendo la duración y el alcance de la señal y las acciones de la insulina. Se han identificado diferentes mecanismos de regulación homeostática a nivel del receptor, del sustrato del receptor de insulina (IRS) y de proteínas localizadas río abajo de ambas, entre las que se encuentran PI3K, Akt o GLUT-4. Diversos estudios han demostrado que la actividad del receptor de insulina es regulada por la acción de fosfatasas de fosfotirosinas que desfosforilan residuos específicos de Tyr del receptor activo, reduciendo de este modo su actividad. En particular, se tiene evidencia de que la fosfatasa de fosfotirosinas 1B (PTP-1B) es un componente esencial de los mecanismos reguladores de las acciones de la insulina. Además de la regulación a nivel del receptor de insulina y del IRS, existen puntos de regulación por debajo de ambas proteínas que también influyen en la modulación de la señal de insulina. En este contexto, las fosfatasas de lípidos pueden regular la señalización de insulina mediante la modulación de los niveles de fosfatidilinositol-3,4,5-trifosfato (PIP3), los cuales se generan por acción de la PI3K. homólogo de la fosfatasa y tensina (PTEN) desfosforila a PIP3, antagonizando así de manera específica la señalización de PI3K/ Akt. Por otra parte, las fosfatasa-2 del inositol en posición 5’ que contiene un dominio SH2 (SHIP-2) también desfosforila a PIP3 y desempeña un papel importante en la regulación de la señal de insulina. 4 La resistencia a la insulina es un estado patológico muy estudiado en la actualidad. Se le atribuye una vital importancia en el estudio de la DM, en el que las células que responden a la insulina dejan de hacerlo. Los individuos con resistencia a la insulina están predispuestos al desarrollo de esta entidad, de forma específica a la diabetes mellitus tipo 2 (DM2), además de asociárseles de forma frecuente con un número importante de desórdenes de salud entre los que se encuentran: la obesidad, la hipertensión, infección crónica y enfermedades de tipo cardiovascular. 3
De manera general, la resistencia a la insulina se manifiesta por una disminución en el transporte de glucosa inducido por la insulina en adipocitos y músculo esquelético, un aumento de la producción de glucosa hepática y alteraciones en el metabolismo de lípidos en tejido adiposo y hepático.
A nivel molecular, los mecanismos por los que se genera la resistencia a la insulina pueden ser múltiples y variar de un individuo a otro. Sin embargo, la resistencia a la insulina es la consecuencia de una deficiente señalización de la insulina causada por mutaciones o modificaciones post consecuencia de una deficiencia del receptor de insulina o de moléculas efectoras río abajo, lo que provoca disturbios en el metabolismo de la glucosa que estimula la aparición de alteraciones metabólicas. Entre las más comunes se encuentran la disminución en el número de receptores y de su actividad de cinasa; un aumento en el estado de fosforilación en residuos de Ser/Thr de proteínas clave como el receptor y su sustrato; la disminución de la actividad de las cinasas PI3K y Akt y defectos en la expresión y función del transportador GLUT4. De estas alteraciones el aumento en la fosforilación en residuos de Ser/Thr a nivel del receptor de insulina y de (IRS), se considera el mecanismo clave en el desarrollo de la resistencia a la insulina. Un aumento en el estado de fosforilación de ambas proteínas puede alterar su asociación a otras proteínas, bloquear sitios de fosforilación en Tyr, disminuir su activación e inducir su degradación. La importancia de un aumento en el estado de fosforilación en residuos de Ser/Thr de las proteínas (IRS) también ha sido documentado en estudios clínicos, en los que se ha demostrado que en el hígado, músculos y tejido adiposo de pacientes obesos (tejidos que desempeñan un papel importante en el desarrollo de la resistencia a la insulina), la expresión de las proteínas IRS-1 disminuye alrededor del 54 % y este aumento en la degradación de (IRS) puede estar dado por un aumento en la fosforilación de (IRS) en residuos de Ser/Thr. (3
Se considera que la resistencia a la insulina se manifiesta cuando el control homeostático es insuficiente quizás debido a defectos en la secreción de la insulina por parte de las células beta donde pueden existir disturbios en los mecanismos moleculares de la acción de la insulina y la regulación de su señal, esto conlleva a niveles elevados de glucosa en sangre lo que favorece la aparición de complicaciones crónicas sobre todo en la DM tipo 2. 2
Existen dos hipótesis que explican su patogenia, la glucotóxica y la lipotóxica. El modelo clásico es la glucotóxica, en este se le otorga a la hiperglicemia y al metabolismo mitocondrial un papel central en las complicaciones asociadas a la enfermedad. 3
Los eventos metabólicos que condicionan la hiperglicemia e influyen en la posterior evolución clínica del paciente denominado memoria metabólica, se pueden generalizar y dentro de estos mecanismos de propagación está la glicosilación no enzimática de proteínas y lípidos celulares. También se incluye el exceso de especies reactivas de oxígeno (ROS) y de nitrógeno, particularmente aquellas originadas en las proteínas mitocondriales glicosiladas, que actúan de forma coordinada para mantener las señales de estrés. 2 Diferentes autores detallan otras hipótesis que intentan explicar las complicaciones asociadas a la hiperglicemia crónica. Dentro de estas se encuentran glicosilación de moléculas, la aldosa reductasa, los trastornos en la actividad de la proteína kinasa C, la pseudohipoxia, el estrés oxidativo, los trastornos del metabolismo de las lipoproteínas y las alteraciones en el funcionamiento de las citoquinas. 4
A continuación, de forma breve, se expresan algunas de estas hipótesis. La teoría molecular de la aldosa reductasa, plantea que el incremento en la vía de los polioles o del sorbitol, en los tejidos que toman libremente la glucosa de la sangre, que no requieren de insulina para su captación y que contienen esta enzima: riñón (podocitos, células mesangiales y epitelio tubular), tejido nervioso (axones y células de Schwann) y tejido ocular (epitelio corneal, cristalino y pericitos retinales), el flujo de este monosacárido al interior de sus células está limitado en condiciones de normoglucemia, tanto por las concentraciones intracelulares de este azúcar como por su poca afinidad con la enzima. 4
Esta vía es una cascada de reacciones químicas en la cual se obtiene fructosa a partir de la glucosa, pasando por el sorbitol, juega un papel protagónico en este proceso la enzima aldosa reductasa. El sorbitol no difunde fácilmente a través de las membranas, por lo que su aumento dentro de la célula contribuye a incrementar la presión osmótica intracelular y los tejidos se dañan por el edema celular. 4
La proteína kinasa C (PKC), importante proteína que participa en las cascadas moleculares antes descritas en el mecanismo molecular de acción de la insulina, pertenece a la familia de las serinas/treoninas fosfocinasas (enzimas con capacidad de fosforilar proteínas), presenta, por lo menos, 11 isoformas codificadas por 10 genes diferentes, de los cuales la b y la d son las que se activan esencialmente por la hiperglucemia. Las alteraciones celulares estructurales y funcionales atribuidas a la activación de la PKC son muy variadas y dependen de la afectación de la función de esta enzima en los mecanismos de transducción de señales y en su participación en la regulación de la expresión de diversos genes, incluyendo a los que codifican la síntesis de proteínas de matriz extracelular (fibronectina y colágeno tipo IV), del inhibidor del activador del plasminógeno (PAI-1) y del factor de crecimiento-transformación b1 (TGF-b1) y su receptor. Como consecuencia de la hiperactivación de PKC se presenta, entre otras, disminución de la producción de óxido nítrico (ON), e incremento de la producción de la endotelina-1, lo cual provoca vasoconstricción e hipoxia tisular, induce la agregación promoviendo la hipertensión y la aterogénesis. La exposición a la glucosa, a los ácidos grasos libres en el tejido arterial induce la producción de aniones superóxido y disminuye la biodisponibilidad del óxido nítrico en la pared vascular, el tratamiento antioxidante actúa recuperando la función endotelial bajo estas condiciones. Una producción normal de especies reactivas de oxígeno ROS es necesaria para el buen funcionamiento de las células endoteliales, pero en la diabetes, un incremento en la producción de estas especies lleva a la disfunción. (5 En este trabajo se expondrá de manera específica las hipótesis vinculadas a la formación y acumulación de productos finales de la glicosilación avanzada y el estrés oxidativo consecuente. Aunque se debe destacar que en el metabolismo todos estos procesos, relacionados con las diferentes hipótesis, están estrechamente vinculados.
Papel del estrés oxidativo
La esencia de esta teoría radica en que las células aeróbicas, al captar la molécula oxígeno (O2) imprescindible para la viabilidad celular por su papel como aceptor final de la cadena transportadora de electrones (CTE) en la respiración mitocondrial, producen una reducción parcial de este oxígeno y además de originar agua (H2O) como producto final, se generan radicales libres (RL) y especies reactivas de oxígeno (ROS). 6-9) Un exceso de radicales libres rompe el equilibrio celular dando lugar al inicio de una serie de reacciones químicas que pueden conducir a la aparición de graves desórdenes fisiológicos y a la agudización de la enfermedad o incluso alterar el desempeño físico o psíquico de una persona supuestamente sana. Las sustancias oxidantes pueden actuar sobre cualquier molécula, aunque algunas parecen ser más susceptibles que otras a la acción de los antioxidantes. Especialmente sensibles resultan los carbohidratos, los ácidos nucleicos, las proteínas y los fosfolípidos presentes en todas las membranas celulares, incluyendo sus posibles derivados. La interacción de los oxidantes con estas moléculas producirá en ellas una modificación estructural, que se traducirá en una alteración funcional. 10,11) La alta inestabilidad atómica de los radicales libres (RL) provoca su colisión con otras biomoléculas a las cuales le sustraen un electrón, oxidándola, perdiendo de esta manera su función específica en la célula. Si se trata de los lípidos (ácidos grasos polinsaturados), se dañan las estructuras ricas en ellas como las membranas celulares y las lipoproteínas. En las primeras se altera la permeabilidad celular conduciendo al edema y la muerte de las células y en la segunda; la oxidación de las lipoproteínas de baja densidad (LDL), lo que conduce a la génesis de la placa ateromatosa. 12) De las proteínas se oxidan preferentemente los aminoácidos (fenilalanina, tirosina, triptófano, histidina y metionina) y como consecuencia se forman entrecruzamientos de cadenas peptídicas, fragmentación de la proteína y formación de grupos carbonilos que impiden el normal desarrollo de sus funciones.13,14 El daño a los ácidos nucleicos, específicamente los ácidos desoxirribonucleicos (DNA) produce bases modificadas, lo que tiene serias consecuencias en el desarrollo de mutaciones y carcinogénesis, por una parte, o la pérdida de expresión por daño al gen específico. Se ha demostrado que la oxidación del ADN mitocondrial se asocia con la oxidación del glutatión mitocondrial. Como consecuencia, la actividad respiratoria global de las mitocondrias disminuye con la edad en el hígado, músculo esquelético y cerebro. Las mitocondrias más envejecidas producen más radicales libres que las jóvenes. En cuanto al ácido ribonucleico (ARN) también es alterado por exposición de la célula a estos radicales. 15-18
Glicosilación no enzimática
La utilización de oxígeno no es el único aspecto del uso de combustibles que puede tener consecuencias lesivas. Algunos combustibles son también moléculas reactivas que pueden producir daño por sí mismas. La más estudiada en este aspecto es la glucosa. Esta molécula, al igual que otros azúcares reductores, puede reaccionar sin necesidad de catalización por enzimas con el grupo amino de las proteínas. Esta reacción produce una base de Schiff que se convierte espontáneamente en producto de Amadori. Sucesivas reacciones de los productos de Amadori y sus derivados con grupos amino de distintas proteínas producen los llamados productos de Maillard, más conocidos como productos terminales de glicosilación avanzada (PTGAs). Esto conlleva a que la glucosa sea el azúcar reductor generalmente considerado en las reacciones de glicosilación no enzimáticas de interés biológico. Sin embargo, cualquier azúcar que posea un grupo carbonilo libre puede reaccionar con los grupos amino primarios de las proteínas para formar bases de Schiff.
La estructura química exacta de estos productos es variada y no completamente conocida, pero al modificar la estructura química de las proteínas modifica de forma evidente la estructura terciaria de estas proteínas y su balance de cargas, lo que modifica las actividades funcionales que dependen de ello, incluyendo actividad enzimática, unión de ligandos, posición en las membranas, así como sus propiedades mecánicas (elasticidad, resistencia a la torsión). En la DM este proceso favorece la aparición de patologías como cataratas, arteriosclerosis, artritis, enfisema y disminución de la función del sistema inmune, asociados normalmente a la edad. Hay que tener en cuenta que frente a la aparición de proteínas conteniendo PTGAs, hay mecanismos que protegen de los daños producidos por ellos, esto explica que la acumulación de PTGAs es la consecuencia de un balance entre su formación y su degradación. Por lo tanto, los daños asociados a la edad pueden deberse a un aumento de formación de proteínas conteniendo PTGAs o a una disminución de su tasa de eliminación. Los macrófagos contienen receptores para proteínas conteniendo PTGAs, lo que permite su eliminación, y a su vez, durante este proceso, el macrófago libera citocinas como factor de necrosis tisular (TNF) e interleucina 1, que colaboran en el control del proceso de reparación tisular. Así, la bien conocida disminución de la función del sistema inmune con la edad podría explicar la aceleración de los daños tisulares inducidos por los PTGAs. Además de su interacción con las proteínas, los azúcares reductores pueden también formar aductos con el ADN, y como consecuencia, producirse enlaces con proteínas. Hasta ahora, la disminución de la actividad biológica como consecuencia directa de la glucosilación se ha observado en un reducido número de casos, incluyendo varios sistemas enzimáticos donde grupos aminos participan en el proceso de catálisis. En este grupo se puede mencionar a la calmodulina, la superóxido dismutasa y la bomba de calcio en eritrocitos humanos. En algunos sistemas enzimáticos como por ejemplo la aldolasa reductasa, se ha visto que por el contrario, la glucosilación produce un aumento de su actividad. Estas modificaciones de la actividad enzimática pueden dar lugar, in vivo, a alteraciones de los procesos celulares en los que participa cada enzima, por ejemplo tenemos la bomba de calcio que interviene en la regulación de la concentración de Ca2+ en el interior de las células. Las variaciones en dicha concentración, inducidas por diversos estímulos están involucradas en los procesos de contracción muscular, expresión génica, diferenciación celular, secreción
y varias funciones neuronales.
La inactivación parcial de esta enzima, producto de la glucosilación no enzimática, podría provocar alteraciones en estos importantes procesos. Por otra parte, la calmodulina ha sido reconocida como el principal mediador de los estímulos regulados por Ca2+ y su alteración traería graves consecuencias al normal funcionamiento de las células. Por otro lado, la superóxido dismutasa que desempeña un papel fundamental en los mecanismos de defensa del organismo frente a los radicales libres de oxígeno, al inhibirse por glucosilación podría incrementar el efecto nocivo de dichos radicales. 4 Recientemente se ha demostrado que los PGA se forman también en las proteínas de la célula in vivo. Un ejemplo de glucosilación avanzada intracelular que merece ser destacado ocurre en los eritrocitos. La albúmina glicada supone el 80 % de las proteínas glicadas circulantes y la albúmina modificada con Amadori es la forma predominante in vivo. En la albúmina in vivo, los principales residuos sujetos a glicación son lisinas en las posiciones 525, 439, 281 y 199, y es la más importante la modificación en la posición 5254. 19 Algunos estudios han demostrado que la albúmina es glucosilada de manera postrancripsional y en abundancia en los individuos diabéticos, por lo que se ha planteado, que la medición de la albúmina glucosilada sería también de valor para evaluar el grado de control metabólico a corto plazo en estos sujetos. Otras proteínas plasmáticas que pueden sufrir el proceso de glucosilación-oxidación, son las lipoproteínas, favorecido por la presencia en estas de ácidos grasos poliinsaturados, que se pueden oxidar con facilidad. Las lipoproteínas glucosiladas, oxidadas y glucooxidadas están implicadas en la patogenia de la enfermedad microvascular y macrovascular en la diabetes mellitus, porque son especialmente aterogénicas, sobre todo, la LDL, la cual ha sido la más estudiada y puede presentar diferentes modificaciones, como un descenso de 25 a 60 % de su contenido de ácido siálico, una disminución de su diámetro y un aumento de su densidad y de su electronegatividad. En el caso particular de las LDL, la glucosilación interfiere en el reconocimiento de esta por su receptor hepático, lo que disminuye su aclaramiento plasmático y aumenta su permanencia en la sangre, situaciones que favorecen que sea captada por la íntima vascular, desde donde migra luego al espacio subendotelial y allí es ingerida por un macrófago, quien no puede metabolizar en su interior el colesterol y se convierte en una célula espumosa, componente fundamental de la placa de ateroma.
La presencia de PTGAs altera las propiedades funcionales de diversos componentes moleculares de la matriz extracelular. El colágeno fue la primera de las proteínas presentes en la matriz en la que se demostró la existencia de enlaces intermoleculares covalentes producidos por los PTGAs, aunque también pueden afectarse la fibronectiva y la laminina, entre otras proteínas. El colágeno es una proteína que por su ubicación está siempre expuesta a la glucosa presente en los líquidos extracelulares, lo que la hace especialmente vulnerable a la glucosilación; se producen entonces modificaciones estructurales que devienen alteraciones de la matriz extracelular. La aparición de estas alteraciones es uno de los mecanismos más importantes implicados en el desarrollo de las complicaciones en la diabetes mellitus (DM). Muchas investigaciones han demostrado que las modificaciones ocurridas en el colágeno de los individuos diabéticos son similares a las que se producen con el envejecimiento del organismo. El colágeno de las personas no diabéticas se glucosila en progresión lineal en función de la edad, pero este fenómeno está muy acelerado en la DM, en la que se pone en marcha un proceso de formación de complejos (agregados) moleculares de entrecruzamiento, que se perpetúan aún en ausencia de glucosa, al estar ya bien establecido. Cuando el colágeno del espacio subendotelial se glucosila, forma productos de entrecruzamiento no solo con otras moléculas de colágeno, sino también con algunas proteínas plasmáticas como la albúmina, las inmunoglobulinas y la LDL. De la acumulación de estas proteínas en el subendotelio, resulta, en parte, el engrosamiento, la disminución de la flexibilidad y el aumento de la permeabilidad que se presentan en las membranas basales capilares afectadas por la glucosilación, además de que puede producirse un estrechamiento de la luz vascular. Se ha precisado que la mielina modificada por el proceso de glucosilación es identificada por determinados macrófagos (carroñeros), que se unen a receptores específicos de PTGAs y entonces la mielina es incorporada al interior de estos macrófagos mediante el proceso de endocitosis, lo que justificaría la presencia de desmielinización segmentaria que se aprecia en los nervios de los individuos diabéticos afectados de neuropatía.
De igual manera, se ha evidenciado que la glicosilación también puede afectar a otras proteínas que componen el citoesqueleto axonal como la tubulina y la actina; esto condiciona un enlentecimiento de la conducción nerviosa y la atrofia y degeneración axonal. Otra proteína que puede sufrir glucosilación es la laminina, lo que provocaría la pérdida de la capacidad de regeneración de las fibras nerviosas en los sujetos diabéticos. La glucosilación del ADN puede ser responsable de numerosos cambios en esta molécula, como su ruptura, deterioro de la reparación, la replicación y la transcripción. Todo lo anterior puede favorecer la aparición de alteraciones cromosómicas y el envejecimiento celular precoz. Se ha postulado que la glucosilación del ADN pudiera ser la causante del aumento de la frecuencia de anomalías congénitas encontradas en los fetos de madres diabéticas y que pueden comprometer a cualquier sistema del organismo. Los PTGAs también pueden interactuar con las histonas, proteínas básicas que constituyen aproximadamente 50 % de la masa total de los cromosomas y que desempeñan un papel importante en el correcto funcionamiento de los genes. 20
Vinculación de la glicosilación no enzimática y el estrés oxidativo
Se conoce que la hiperglucemia, que representa la característica fundamental de la DM, promueve la activación de varias reacciones involucradas en procesos metabólicos donde se generan metabolitos intermediarios que tienen acción prooxidante. Esta también condiciona el aumento del metabolismo anaerobio (glucólisis anaerobia), con la consiguiente excesiva producción y acumulación de lactato, lo que constituye un evento generador de radicales libres. Se sabe que una posible fuente de radicales libres en la diabetes es la autoxidación de la glucosa, la cual facilita la generación de cetoaldehídos reactivos y la formación de PFGA, potentes reductores que generan radicales de oxígeno en presencia de hierro o cobre. Además, el aumento de la oxidación del sorbitol con la consecuente formación de fructosa, puede incrementar la producción del anión superóxido a través de la reducción de la prostaglandina G2 en H2. Por otro lado, la hiperglucemia puede debilitar las defensas antioxidantes, esto facilita que los radicales libres dañen a varias moléculas intracelulares y extracelulares, e incluso, a proteínas estructurales. Así, se ha comprobado una reducción de la concentración de vitamina C en leucocitos y una depleción plasmática de vitamina E en sujetos diabéticos, además de una disminución eritroleucocitaria de enzimas antioxidantes como la superóxido dismutasa, la catalasa y la glutatión peroxidasa. El óxido nítrico (ON) es una molécula gaseosa producida por el endotelio vascular a partir de la L-arginina, y que tiene una potente acción vasodilatadora, además de antiproliferativa y antiagregante plaquetaria. Posee una vida media in vivo muy corta y su síntesis es catalizada por la acción de la enzima ON-sintetasa, de la cual existen dos isoformas: la constitutiva, dependiente de calcio y poco productiva y la inducible por endotoxinas y citoquinas, e independiente de calcio. Se ha demostrado que los PTGAs ligados a proteínas y formados en la matriz vascular pueden reaccionar con el ON e inactivarlo; con ello ocasionan un defecto en la relajación vascular, lo que pudiera explicar, en parte, la disminución de la respuesta vasodilatadora que se observa en las arterias de los individuos con DM y la frecuente aparición de hipertensión arterial que se constata en estos sujetos. Asimismo, se ha comprobado que el defecto en la respuesta vasodilatadora mediada por el ON en la DM, se correlaciona con el nivel acumulado de PFGA y puede evitarse mediante la inhibición de la formación de PFGA. La acelerada inactivación del ON, como consecuencia de su interacción con los PTGAs, conlleva al obligado incremento de la actividad de la ON-sintetasa, lo que aumenta la producción del anión superóxido, el cual interviene en la formación del compuesto proxidante peroxinitrito. Este último compuesto es capaz de oxidar a las LDL e inactivar a la prostaciclina sintetasa, que provoca con ello, finalmente, disfunción endotelial. Debido a que el peroxinitrito es un compuesto difícil de determinar, se usa la medición de su derivado directo, nitrosamina, como indicador de su producción. En la DM está aumentada la actividad de la enzima aldosa reductasa, lo cual produce un gran consumo de NADPH y queda muy escasa cantidad de este disponible para poder ser utilizado por otras enzimas, como la glutatión peroxidasa que tiene actividad antioxidante, fenómeno que pudiera contribuir también al aumento del estrés oxidativo. (20
Recientemente la evidencia experimental sugiere que la glucosilación y el estrés oxidativo se pueden vincular a la vía del sorbitol potenciándose y contribuyendo así al desarrollo de complicaciones diabéticas, aquí se pueden observar los vínculos entre las diferentes hipótesis que se plantean para explicar las complicaciones de la entidad estudiada. Debe ser señalado que la fructosa producida por la vía del sorbitol es extremadamente potente como agente de glucosilación, superando a la glucosa. En el retículo endoplásmico ER, un organelo celular, se realizan importantes funciones celulares, como el almacenamiento del calcio intracelular, el ensamblaje y plegamiento de proteínas y modificaciones postraduccionales. En condiciones de estrés celular, que incrementa la demanda del ER y conlleva una sobrecarga de su capacidad funcional, se generan alteraciones en su función y en la disminución del transporte de proteínas hacia el aparato de Golgi, la expresión de proteínas mal plegadas y la depleción del calcio de este reservorio, que en su conjunto reciben el nombre de “estrés del retículo”. 19 Como mecanismo compensatorio al estrés del ER y de forma específica al mal plegamiento de las proteínas, se activa en el mismo organelo, un mecanismo conocido como respuesta a las proteínas mal plegadas (UPR), (unfolding protein response) (por sus siglas en inglés) que permite restablecer la homeostasis de las funciones del ER, mediante la inhibición de la síntesis de proteínas y el aumento tanto de la degradación de las proteínas del ER como del nivel de chaperonas para ayudar en el plegamiento proteico. Si estos mecanismos de adaptación son insuficientes para restaurar la homeostasis del ER, la célula experimenta la muerte celular programada. (19 La mitocondria, un organelo celular que juega un papel importante en el metabolismo energético, se encarga de proporcionar la mayor parte de la energía necesaria en forma de trifosfato de adenosina (ATP), a partir de moléculas orgánicas que son oxidadas en presencia de oxígeno. 4-21 En los últimos años se ha propuesto que las alteraciones a nivel mitocondrial se asocian a la resistencia a la insulina, pero los mecanismos por los que el daño o disfunción mitocondrial conlleva la resistencia son causa de debate. Por otra parte, existe evidencia experimental cada vez más convincente de que las especies reactivas de oxígeno generadas por la mitocondria tienen un papel importante en la patogénesis, progresión y complicaciones a largo plazo de la DM tipo 2. Se ha propuesto que la disminución en la oxidación de sustratos afecta al flujo de electrones a través de su cadena de transporte, lo que provoca una fuga de estos hacia el oxígeno, formando aniones superóxido. La generación de las especies reactivas de oxígeno provoca diversos daños a nivel mitocondrial y celular, entre los que se encuentran el daño oxidativo en el ADN mitocondrial, la agregación de proteínas, que se glicosilan por los altos niveles de glucosa existentes y la existencia de peroxidación lipídica, todo lo que de forma potencial da lugar a mitofagia. 4,21-23) Es determinante que la memoria metabólica que aparece sobre todo en la DM tipo 2 sugiere la necesidad de un tratamiento enérgico temprano destinado a normalizar el control metabólico para reducir los efectos nocivos de las proteínas glucosiladas y disminuir las especies reactivas, las cuales tiene un impacto sobre el DNA mitocondrial, lo que genera cambios epigenéticos que se podrían perpetuar en las generaciones futuras. 2-24
Por tanto, al existir alteraciones en los mecanismos involucrados en regular los mecanismos moleculares de acción de la insulina y aumentar los niveles de glucosa en sangre, se producen modificaciones que se traducen en la existencia de procesos de glicosilación no enzimática que concomitan con la aparición de radicales libres potencialmente dañinos. (Fig.1).
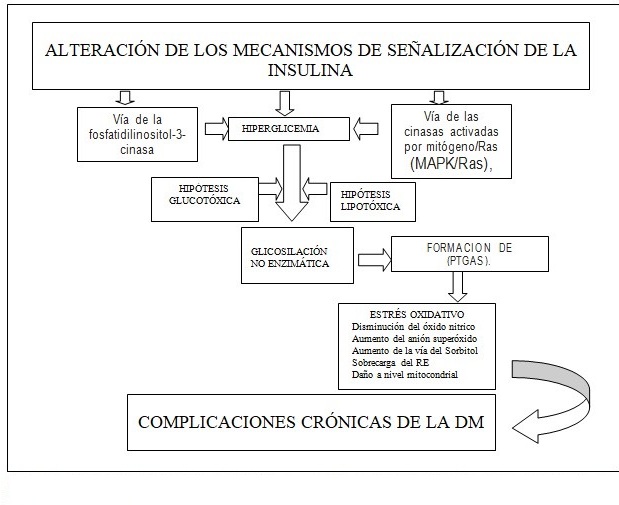
Conclusiones
En la presente revisión se han abordado diferentes aspectos relacionados con la función y señalización de la insulina como hormona vital relacionada con la DM, donde aparecen por diversas causas modificaciones en estos mecanismos moleculares lo que provoca niveles elevados de glucosa en sangre. Se destaca cómo, debido a esta consecuencia resultante, se produce el proceso de glucosilación no enzimática, lo que unido al incremento del estrés oxidativo que la acompaña, puede afectar por sí misma tanto a los carbohidratos, las proteínas, los lípidos y al ADN del organismo. Si se profundiza en estos aspectos se obtiene una mayor comprensión de los mecanismos moleculares de acción hormonal y su desregulación, así como la interconexión que existe entre todos estos procesos metabólicos resultantes que constituyen causa y consecuencia. Todo ello condiciona el estudio de la resistencia a la insulina como un proceso patológico muy estudiado en los momentos actuales y a la disminución de las complicaciones crónicas de esta entidad, lo que constituye un reto para la investigación científica.

