










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El aislamiento del ácido desoxirribonucleico (ADN) resulta indispensable y constituye el primer paso en la mayoría de los procesos biológico-moleculares antes de realizar procedimientos como la secuenciación, amplificación o detección.1 Las técnicas tradicionales de separación utilizan solventes orgánicos tóxicos y procedimientos laboriosos basados en sucesivas extracciones y centrifugaciones que limitan la pureza de los productos de separación, no permiten su automatización y escalado, y someten al ADN a alto estrés mecánico.2 Sin embargo, el uso de tecnologías emergentes como la separación magnética, la cual utiliza los principios del magnetismo para el aislamiento eficiente de partículas magnéticas de suspensiones biológicas,3,4 ofrece una serie de ventajas entre las que destacan el reducido tiempo de procesamiento, la ausencia de reactivos químicos tóxicos, la facilidad en la separación y automatización, además de la separación de ADN de contaminantes y desechos celulares.5,6
Entre las diferentes partículas magnéticas utilizadas en aplicaciones biomédicas, las nanopartículas magnéticas de óxido de hierro (IONPs) resultan de especial interés debido a su baja toxicidad y alta biocompatibilidad.7,8 Las mismas pueden estar compuestas por magnetita (Fe3O4) o maghemita (γ-Fe2O3), sustancias que exhiben altos valores de área superficial, una elevada relación superficie-volumen y propiedades magnéticas que permiten su fácil separación del medio en presencia de un campo magnético y las convierten en materiales ideales para este tipo de procedimiento.9,11 En la obtención de las IONPs sobresale por su simplicidad, reproducibilidad y bajo costo el método de co-precipitación propuesto por Massart.12 Dicho método utiliza disoluciones de Fe2+ y Fe3+ en un medio alcalino en presencia de surfactantes,13 además, de que permite la introducción de sales de otros metales (M) para formar las ferritas correspondientes (MFe2O4). A pesar de su versatilidad, el método no permite un control adecuado de la morfología y tamaño de partículas de los óxidos obtenidos; por lo que obtener resultados reproducibles resulta un reto para los investigadores.14 No obstante, un control estricto del pH y la temperatura durante la síntesis posibilitaría la obtención de partículas con un rango de tamaños estrecho y, por consiguiente, una respuesta magnética homogénea de las partículas.
Usualmente las nanopartículas suelen recubrirse con polímeros, agentes surfactantes, ligantes o materiales inorgánicos (sílice, carbono, metales preciosos u óxidos) que las protegen de la oxidación, las estabilizan o dispersan y le dan, eventualmente, la posibilidad de ser funcionalizadas para enlazarlas con un determinado ente químico o biológico.15 Entre estos métodos, los recubrimientos con sílice (SiO2) se han convertido en la actualidad en una promisoria e importante vía para el desarrollo de nanopartículas magnéticas recubiertas, para aplicaciones biomédicas debido a biocompatibilidad, estabilidad, fácil conjugación con diferentes grupos funcionales y posibilidad de acoplarse a dianas biológicas con selectividad y especificidad.16,17 Por otro lado, dichos recubrimientos potencian la habilidad de formación de un enlace entre la superficie de las partículas magnéticas y el ADN (figura 1) debido a la introducción de una pequeña carga iónica positiva que evita la agregación de las partículas magnéticas y facilita la interacción con este.18
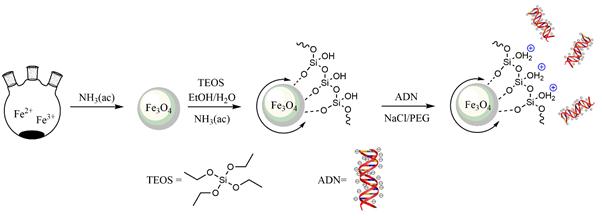
Todas las estrategias de protección resultan en nanopartículas magnéticas con una estructura núcleo-coraza, lo que podría atenuar la fuerza magnética del núcleo; sin embargo, el control estricto del espesor del recubrimiento posibilita obtener nanopartículas de tipo Fe3O4@SiO2 con características adecuadas para las aplicaciones deseadas.
Las rutas sintéticas para obtener Fe3O4@SiO2 pueden ser divididas en tres categorías: i) materiales magnéticos en el interior de matrices de sílice pre-sintetizadas;19 ii) partículas magnéticas con estructura núcleo-coraza fabricadas in situ;20 y iii) recubrimientos de sílice en la superficie de partículas magnéticas previamente sintetizadas.21 Siendo este último el más eficiente en la obtención de partículas Fe3O4@SiO2. En esta última variante destaca entre los más populares el método de Stöber o modificaciones de este,22,26 el cual permite la obtención de partículas homogéneas mediante un procedimiento simple que involucra la hidrólisis y policondensación del tetraetilortosilicato ENT#091;(C2H5O)4 Si, TEOSENT#093; bajo condiciones alcalinas en etanol y a temperaturas superiores a 60°C, además del control de la espesura de los recubrimientos.27
Por tal motivo el objetivo principal de nuestro trabajo fue la síntesis y caracterización de nanopartículas magnéticas de óxidos de hierro recubiertas con sílice, para su posible aplicación en la separación de ADN, péptidos o biomoléculas.
Materiales y métodos
Obtención de las magnetitas
La síntesis de la magnetita se llevó a cabo por el método de co-precipitación descrito por Massart 12) en el cual se ponen a reaccionar dos soluciones de Fe2+ y Fe3+ en medio básico en una proporción de 1:2. Previamente se mezclan 5 mL de FeCl2.4H2O (2 mol/L) y 20 mL de FeCl3.6H2O (1 mol/L) obtenidas por disolución de las sales en HCl (2 mol/L). En un balón de tres bocas se colocan 250 mL de NH3 0,7 mol/L y se gotea poco a poco (1 mL/min) la disolución de las sales de Fe2+/Fe3+ bajo agitación mecánica constante (500 rpm). La reacción se lleva a cabo en un sistema cerrado a temperatura ambiente y en atmósfera de nitrógeno, lo cual permite desplazar el oxígeno presente en el sistema y evitar la oxidación del hierro (II) a hierro (III). Una vez concluido el goteo y después de la aparición de un precipitado negro las partículas magnéticas se dejan sedimentar por 20 min, se decanta el líquido sobrenadante con la ayuda de un imán y se lavan. Finalmente, las partículas se secan en estufa a 60 °C y se guardan en un recipiente cerrado para su posterior recubrimiento.
La tabla 1 representa las condiciones de síntesis en las que se llevaron a cabo los diferentes experimentos y la leyenda utilizada para nombrar las muestras luego de su recubrimiento.
Tabla 1 Condiciones experimentales durante la síntesis de las magnetitas
| Muestras Fe3O4@SiO2 | Condiciones de síntesis | |||
|---|---|---|---|---|
| Compensación de pH | Lavado | |||
| Sí (pH final=12) | No (pH final=9) | H2O | HClO4 | |
| (T1) | X | X | ||
| (T2) | X | X | ||
| (T3) | X | X | ||
| (T4) | X | X | ||
Las magnetitas se sintetizaron en condiciones normales y con compensación de pH. En este último caso, durante el experimento se goteó NH3 concentrado a una velocidad de 1 mL/min. La cantidad adicionada se calculó previamente en base al consumo estequiométrico de este reactivo durante la formación de la magnetita y a la cantidad necesaria para la neutralización de la solución ácida de Fe2+/Fe3+. Adicionalmente se varió la disolución de lavado utilizando agua destilada o HClO4 (2 mol/L). En el primer caso se lavó hasta la neutralización de las aguas de lavado y en el segundo se lavó con agua luego del HClO4 igualmente hasta pH=7. Al finalizar, en ambos casos se extrajo el agua remanente mediante lavados con etanol.
Obtención de las Fe3O4@SiO2
Los recubrimientos de las magnetitas fueron realizados por el método de Stöber utilizando TEOS como fuente de silicio (Si) y de acuerdo a la metodología propuesta por Quy y col.28 Para ello, se prepararon 100 mL de una suspensión acuosa a partir de 1 g de magnetita y se mezclaron con 50 mL de una disolución etanólica al 10 % en TEOS. Posteriormente se ajustó el pH de la disolución con NH3 y se colocó en un baño de aceite a 90 °C bajo agitación mecánica constante (500 rpm) por un período de 6 h. Al concluir la reacción se enfrió el recipiente a temperatura ambiente, se decantó la disolución sobrenadante con ayuda de un imán y las magnetitas recubiertas se lavaron con etanol absoluto y acetona.
Para realizar los experimentos posteriores de aislamiento de ADN, las Fe3O4@SiO2 se almacenaron en dos disoluciones buffer compuestas por una disolución 100 mM TRIS-HCl (una a pH=5 y la otra a pH=7) de tal forma que la concentración másica fuese igual a 25 mg/mL.
Purificación de ADN plasmídico
Incubación y lisis celular de Escherichia coli
El plásmido utilizado para la purificación del ADN fue el pcDNA3.1 Zeo(+) Venus-1 Zipper donado por Michnick y colaboradores.29 La cepa de E. coli DH-10B transformada con el plásmido fue inoculada en 5 mL de medio Luria Broth suplementado con Ampicillina (50 μg/mL (LBA), dejándose crecer durante 16 h a 37 °C. Una vez crecidos, se llevó a cabo la lisis de los cultivos utilizando el método modificado de lisis alcalina. Para ello los cultivos se centrifugaron a 10000 rpm durante 5 min y los precipitados se re-suspendieron en 200 µL de solución P1 (50 mM Tris-HCl, pH = 8,0, 10 mM EDTA, 100 μg/mL RNasa A), incubándose 5 min a temperatura ambiente. Posteriormente, se añadieron 400 µL de solución alcalina P2 (NaOH 0,2M/SDS 1 %) y se adicionaron 300 µL de una disolución 7,5 M de NH4C2H3O2 y pH = 7,8 al lisado celular. La muestra se agitó por inversión para luego incubar en hielo durante 20min. Se centrifugó a 10000 rpm por 10 min y se extrajo el sobrenadante de las muestras para la purificación del ADN.30 El sobrenadante de la lisis alcalina se utilizó para la purificación del ADN por precipitación etanólica, mediante columnas de QIAprep® y utilizando las partículas de Fe3O4@SiO2 como soporte sólido.
Purificación alcalina de ADN plasmídico mediante columnas de intercambio iónico marca comercial QIAGEN
Para la purificación del ADN plasmídico se utilizó el kit comercial QIAprep® Spin Miniprep Kit (QIAGEN, Alemania) y se siguieron las recomendaciones del fabricante. Se aplicó el sobrenadante de la lisis alcalina a la columna de centrifugación QIAprep 2.0. La muestra se centrifugó a 10000 rpm por 30-60 s y la columna se lavó agregando 0,75 mL de buffer de lavado (PE): 10 mM Tris-HCl, pH = 7,5, 80 % etanol seguido de una centrifugación a 10000 rpm durante 1 min. Una vez centrifugada, se colocó la columna QIAprep 2.0 en un tubo de microcentrífuga limpio de 1,5 mL y se añadieron 50 μL buffer EB al centro de la columna de centrifugación; dejándola reposar durante 1 min para la posterior elución del ADN mediante centrifugación a 10000 rpm por 1 min.
Purificación del ADN plasmídico por precipitación etanólica 31
Al sobrenadante de la lisis alcalina se le añadieron 450 µL de 2-propanol y luego se centrifugó a 10000 rpm por 10 min. El ácido nucleico precipitado se lavó con 100 µL de etanol al 70 % y nuevamente se centrifugó por 5min. El ADN precipitado se secó por 2-5 min y posteriormente se re-suspendió en 50 µL del buffer de elusión (EB): 10 mM Tris-HCl, pH = 8,5, 1 mM EDTA para su posterior análisis.
Purificación de ADN plasmídico utilizando Fe3O4@SiO2 como soporte sólido
Al sobrenadante de la lisis alcalina se le añadieron 100 µL de la suspensión de Fe3O4@SiO2 y se mezcló por agitación durante 1 min en vórtex. A esta mezcla se le adicionaron 600 µL de un buffer de enlace (BB) preparado a partir de NaCl 1,25 M y 10 % de polietilenglicol (6000). 32 Se mezcló por inversión y se incubó a temperatura ambiente por 1 min colocándolo a continuación en el separador magnético durante 2 min y eliminando el sobrenadante por inversión. Una vez inmovilizado el ADN se lavó con 100 μL del buffer de lavado (30 mM TRIS-HCl pH=7, 2,25 M GUSCN, 56 % etanol) y se colocó una vez más en el separador magnético por 1 min. Una vez pasado este tiempo se desechó el exceso cuidadosamente y las perlas magnéticas y el ADN inmovilizado se lavaron por 30 s con 200 μL de etanol al 70 % y posteriormente fueron secadas al aire durante 2-5 min. Para liberar el ADN de las Fe3O4@SiO2 se adicionaron 100 μL del EB previamente calentado a 65 °C y ambos se mezclaron mediante agitación en vórtex por 1 min. A continuación, los tubos se colocaron en el separador magnético por 1 min y se transfirieron 50 μL del sobrenadante con el ADN purificado a un nuevo recipiente para la cuantificación.
Cuantificación y análisis del ADN recuperado
Para verificar la concentración y la calidad de las muestras de ADN plasmídico se utilizó el espectrofotómetro Nanodrop® ND-1000 (Thermo Scientific, EUA) y se realizó visualización del ADN mediante la electroforesis en gel de agarosa al 0,8 %.
Caracterización de las Fe3O4@SiO2
Fluorescencia de Rayos X (FRX)
Se utilizó la técnica de Fluorescencia de Rayos X como método cualitativo en la identificación de Si en las magnetitas recubiertas. Los ensayos se llevaron a cabo en un equipo de la OXFORD Instruments, modelo X-Supreme utilizando 5 kV de aceleración, una corriente de 400 mA y un tiempo de adquisición de 47 s. Solo se analizó la presencia o no del pico de emisión del Si antes de analizar las muestras con otras técnicas de caracterización.
Espectroscopia IR con transformada de Fourier (FTIR)
Los espectros infrarrojos de las IONPs (con y sin recubrimientos) fueron obtenidos en un equipo SHIMADZU-IR Prestige 21/AIM 8800 en modo ATR (Reflexión Total Atenuada) en un rango espectral de 500-4000 cm-1.
Difracción de rayos X (DRX)
En la obtención de los difractogramas fue empleado un difractómetro SHIMADZU XRD-7000 con radiación de CuKα (λ = 1,540 6 Å) y filtro de Ni. Las mediciones fueron realizadas con un paso angular de 0,025°, tiempo de conteo 5 s e intervalo angular de 25-65 en 2θ. Para el cálculo del tamaño de cristalita se empleó el método de Rietveld 33 asociado a los softwares FullProf y WINPLOTR, el cual emplea el procedimiento de mínimos cuadrados para ajustar y comparar los parámetros estructurales experimentales con los calculadas en un modelo estructural teórico.
Resultados y discusión
Caracterización estructural de las nanopartículas Fe3O4@SiO2
En la figura 2 pueden observarse los espectros de FRX de la magnetita y de las muestras recubiertas bajo las diferentes condiciones de síntesis. En todas las muestras, se analizó la línea de emisión de mayor intensidad del Si (K(1) en 1,739 keV, aun cuando la cercanía a la línea de emisión del tubo de Rayos X (M(1 1,785 9 keV) podría dificultar el análisis.
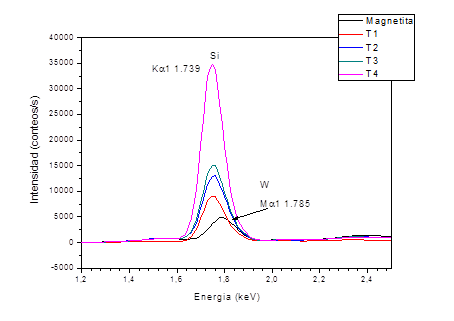
Al comparar los espectros de las muestras recubiertas con el de la magnetita se observaron diferencias en los espectros de emisión, indicando la presencia de Si en todas las muestras y la formación de las Fe3O4@SiO2. Las muestras T3 y T4 poseen una mayor cantidad de Si adsorbido en comparación con T1 y T2, las cuales fueron estabilizadas con HClO4 antes del recubrimiento. La muestra T4 revela una cantidad de Si significativamente mayor que el resto de los especímenes probablemente debido a la compensación del pH durante la síntesis que produce la disminución del tamaño de partícula y, por consiguiente, el aumento del área superficial de las IONPs favoreciendo así la quimisorción del Si. La poca adsorción de Si en T1 y T2 podría estar relacionada con la carga positiva superficial que adquieren las magnetitas al ser estabilizadas en medio ácido, lo cual provoca que durante el proceso de recubrimiento sea necesaria la desprotonación de la superficie antes de que ocurra la policondensación del TEOS sobre la misma;34 siendo este factor determinante en el caso de la muestra T2, también obtenida con compensación de pH.
La figura 3 muestra los espectros infrarrojos de la magnetita y de las Fe3O4@SiO2. En todas las muestras pueden observarse la presencia de una banda sobre los 3500 cm-1 correspondiente a la vibración ν 1 del enlace O-H y otra en 1637 cm-1 correspondiente al doblaje del enlace H-O-H. Dichas bandas se deben a la presencia de humedad en la muestra de magnetita analizada, ya sea por un secado incompleto o por la alta humedad atmosférica durante el análisis.
Usualmente, los espectros de magnetita muestran cuatro bandas características a bajas frecuencias, debido a la estructura de espinela inversa donde pueden observarse las vibraciones de valencia del enlace Fe-O. La frecuencia a la que aparecen dichas bandas depende de la posición del catión en la estructura; es decir si este se encuentra ocupando sitios octaédricos (MO) o tetraédricos (MT). Las mismas pueden encontrarse en los rangos siguientes MT-O-MO (ν 1 ~ 600-550 cm−1), MO-O (ν 2 ~ 470 cm−1), MT-MO (ν 3 ~ 350-400 cm−1) y OT-M-OO (ν 4 ~178 cm-l). (9 En nuestro caso, debido a las limitaciones instrumentales solamente es posible observar la presencia de las vibraciones de valencia (1 correspondientes a la interacción MT-O-MO a una frecuencia de 590 cm−1. Dicha banda aparece ligeramente ensanchada, lo que pudiera indicar una mínima y parcial oxidación de la magnetita a maghemita, ya que algunos autores reportan que en la magnetita pura esta banda es estrecha y con la transformación de una fase en otra inicialmente se ensancha para luego desdoblarse en dos bandas que aparecen en 630 cm−1 y 430 cm−1, debido a cambios en la simetría de la espinela inversa y la aparición del incremento en vacancias de cationes.35
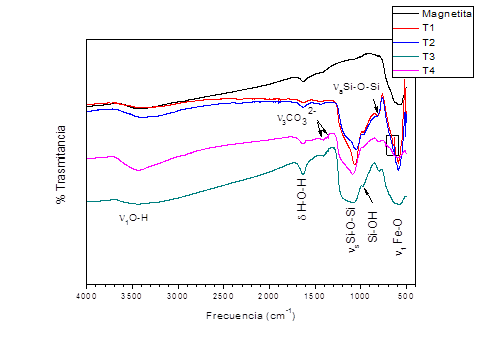
En el caso de las muestras recubiertas, se observan también las bandas correspondientes a las vibraciones simétrica y antisimétrica de los enlaces Si-O-Si a 1080 cm-1 y 800 cm-1 respectivamente y a los enlaces Si-OH (~985 cm-1); los cuales aparecen como un pequeño hombro superpuesto a la (s Si-O-Si. No es posible observar con claridad la banda correspondiente a la vibración Fe-O-Si debido a que esta aparece a bajas frecuencias y en algunos casos pueda estar sobrelapada con la de los grupos Fe-O. Sin embargo, en las muestras T1 y T2 puede observarse aproximadamente a 640cm-1 un hombro que pudiera asociarse a esa vibración. Adicionalmente, puede observarse en todos los recubrimientos, la presencia de un doblete a 1350 cm-1 y 1400 cm-1 correspondiente a la vibración de valencia del ion CO3 2-, probablemente debido a la disolución del CO2 atmosférico durante el proceso de síntesis.
Los espectros obtenidos son similares a los reportados por otros autores para nanopartículas magnéticas obtenidas por co-precipitación inversa in situ de magnetita en presencia de Na2SiO3, sugiriendo que el método de recubrimiento fue efectivo y que la sílice se encuentra quimiadsorbida sobre las partículas de magnetita.36
En la figura 4 pueden observarse los difractogramas para la magnetita y las muestras T1, T2, T3 y T4. La magnetita y la maghemita cristalizan en el mismo sistema cristalino (FCC) y poseen patrones de difracción muy similares, exceptuando la posible presencia de los planos (2 1 0), (2 1 1) y (4 2 1) en esta última fase cuando ocurre el ordenamiento de vacancias. Por tal motivo, usualmente es necesario el uso de técnicas complementarias para la determinación de este último óxido. En todas las muestras analizadas se observó la presencia de Fe3O4 (magnetita) JCPDS 19-0626 como única fase y no se observaron picos que indicaran la presencia de maghemita.37 No fue posible determinar la presencia de silicio por esta técnica, debido al carácter amorfo de dicha sustancia y al bajo contenido en las muestras, posiblemente cercano al límite de detección.
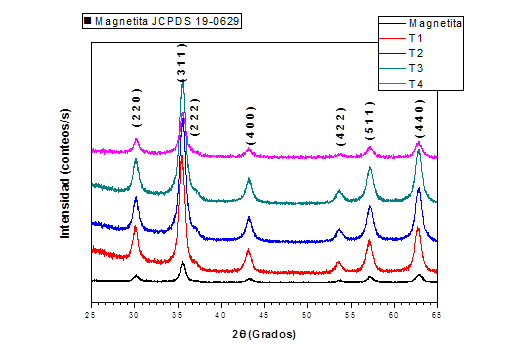
En la tabla 2 se presentan los datos de tamaño de cristalita y parámetro de la celda unitaria (a) luego del ajuste mediante Rietveld (figura 5) y determinados a partir de la línea de difracción más intensa (2( = 35,423) correspondiente al plano (3 1 1). En todas las muestras el parámetro de la celda unitaria presenta un valor intermedio entre los valores teóricos de ambos óxidos de hierro, lo cual puede deberse a: i) decrecimiento del valor de este parámetro con la disminución del tamaño de partícula; ii) presencia de defectos estructurales o iii) posible oxidación parcial de la magnetita a maghemita.38 En este caso, los resultados sugieren que la disminución del parámetro está asociada con la disminución del tamaño de partícula y el posible aumento de defectos estructurales asociadas a este fenómeno y no a la presencia de maghemita.
Tabla 2 Tamaño de cristalita, parámetro de la celda unitaria (a) y parámetro residual obtenidos por el método de Rietveld
| Muestras | Parámetro de la celda a (Å) | Tamaño de cristalitas (nm) | Parámetro Residual (S) |
| Fe3O4@SiO2
|
8,359 | 9,0 | 1,32 |
| Fe3O4@SiO2
|
8,359 | 8,2 | 1,21 |
| Fe3O4@SiO2
|
8,360 | 10,8 | 1,41 |
| Fe3O4@SiO2
|
8,371 | 11,7 | 1,18 |
| Magnetita |
8,396 | -- | -- |
| Maghemita |
8,347 | -- | -- |
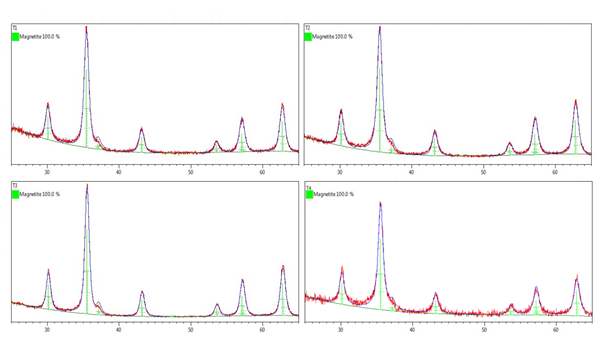
Fig. 5 Difractogramas obtenidos luego del ajuste mediante el método de Rietveld. Línea roja representa la data observada y la línea azul representa el modelo teórico ajustado
La muestra T2 muestra el menor valor de tamaño de cristalita de toda la serie seguida por T1, T3 y T4. Como es conocido, el tamaño de las cristalitas aumenta al disminuir el pH y la fuerza iónica del medio.39 Durante el proceso de síntesis de la magnetita, el pH del medio va disminuyendo a medida que la reacción tiene lugar y debido a la neutralización del HCl proveniente de las disoluciones Fe2+/Fe3+; de manera que cada nueva gota de esta disolución al caer en el medio encuentra un pH menor y las partículas de magnetita que se forman son cada vez mayores obteniéndose así una distribución asimétrica de tamaño de partículas.40 Cuando se realiza la síntesis con compensación de pH, se adiciona simultáneamente la misma cantidad de OH- que se consume en el proceso, aumentando así el pH hasta un valor final de 12, lo cual explica que el valor de tamaño de cristalina de la muestra T2, obtenida en estas condiciones, sea menor que las obtenidas sin compensación de pH. Esto debiera ocurrir igualmente para la muestra T4, aunque en este caso el mayor tamaño de cristalita se debe a la cantidad de Si adsorbida durante el recubrimiento.
Por otro lado, la estabilidad de las suspensiones magnéticas resulta del equilibrio entre las fuerzas repulsivas y atractivas que se establecen entre las partículas magnéticas. Teóricamente existen cuatro tipos de fuerzas que pueden contribuir a la estabilidad de las mismas; sin embargo, las de mayor interés son las interacciones electrostáticas y las repulsiones estéricas. En los óxidos de hierro la superficie actúa como un ácido de Lewis coordinándose con moléculas de agua que donan pares electrónicos libres para posteriormente disociarse, dejando la superficie de las nanopartículas de hierro funcionalizadas con grupos hidroxilos anfotéricos. Dichos grupos OH a su vez actúan como ácidos o bases dependiendo del pH del medio: a pH>7 los IONPs se cargan negativamente, mientras que a pH<7, como el que se produce al utilizar HClO4, las nanopartículas se cargan positivamente creando una repulsión electrostática que previene la aglutinación de las partículas.41
Al comparar las muestras sintetizadas en las mismas condiciones, aquellas muestras en las que se utilizó HClO4 en lugar de H2O presentan menores tamaños de cristalita, aunque el factor determinante en este parámetro parece ser la compensación del pH. De acuerdo con los resultados obtenidos el tamaño de partícula debe seguir el siguiente orden T2<T4<T1<T3. Sin embargo, la muestra T4 es la de mayor tamaño de cristalita debido a la mayor cantidad de Si adsorbido sobre la superficie de la muestra de acuerdo a los datos de FRX (figura 1).
Los valores de tamaño de cristalita de las Fe3O4@SiO2 obtenidas son similares a los obtenidos por otros autores para magnetitas obtenidas por co-precipitación y funcionalizadas con SiO2, quitosana, APTES y TRIS.42,43 Además, se encuentran por debajo del valor crítico de 20nm que reporta la literatura para el cual es observado el fenómeno del superparamagnetismo 44 haciéndolas ideales para aplicaciones biomédicas.
Purificación de ADN plasmídico utilizando Fe3O4@SiO2 como soporte sólido
El método de extracción de ADN ideal debe ajustarse a los siguientes criterios: ser sensible, consistente, rápido y fácil de usar, y dependiendo del país en el que se utilice, puede ser importante minimizar el equipo especializado o el conocimiento bioquímico. También debe suponer un riesgo mínimo para los usuarios, así como evitar la posible contaminación cruzada de las muestras.45 Finalmente, y lo más importante, la técnica de extracción de ADN elegida debería ser capaz de entregar muestras de ADN puro listas para ser utilizadas en aplicaciones moleculares. En la figura 6 se muestran los resultados de la electroforesis en gel agarosa de ADN del plasmídico pcDNA3.1 Zeo(+) Venus-1 Zipper de E. coli purificado por tres metodologías. Como controles positivos se utilizaron el método de lisis alcalina seguido de precipitación etanólica (linea1) y el sistema comercial QIAprep Spin Miniprep Kit (línea 2) y se comparó con las cuatro muestras de nanopartículas de Fe3O4@SiO2 T1, T2, T3 y T4 (líneas 3-6). En este caso, sólo la muestra T3 mostró resultados comparables a los controles positivos y no se observaron diferencias en la eficiencia de la separación de acuerdo a los resultados de la electroforesis. Este hecho es posible explicarlo teniendo en cuenta que, durante la experimentación, las nanopartículas correspondientes a la muestra T3 fueron las que mejor se resuspendieron a pH=5. Las muestras T1 y T2 no se unieron de manera eficiente al ADN plasmídico no permitiendo así su purificación, posiblemente debido a la poca cantidad de silicio que fue encontrada en dichas muestras (figura 2). La muestra T4 tampoco permitió la purificación del ADN y no se encontró ninguna evidencia teórica o experimental que pudiera fundamentar este resultado.
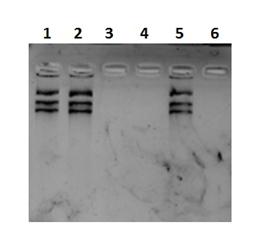
Fig. 6 Electroforesis en gel de Agarosa del ADN plasmídico pcDNA3.1 Zeo(+) Venus-1 Zipper. Línea 1 representa el ADN obtenido por precipitación etanólica, Línea 2: Columna de QIAprep® y Línea 3 a 6: muestras de nanopartículas de Fe3O4@SiO2 T1, T2, T3 y T4
La medición de la absorbancia en la región ultravioleta del espectro visible usando espectrofotometría a diferentes longitudes de onda (230 nm, 260 nm y 280 nm) es una forma inicial rápida y eficiente de determinar la pureza y la concentración de muestras de ácido nucleico purificado (tabla 3). La concentración del ADN recuperado generalmente se calcula a partir de la lectura de la absorbancia a 260 nm; mientras que la pureza se evalúa de acuerdo a la relación de absorbancia de 260/280, siendo aceptables valores en el rango de 1,8-2,0 los cuales denotan ausencia de contaminantes proteicos. Las relaciones de absorbancia 260/230 entre 2,0 y 2,2 también se consideran adecuadas como medida secundaria de pureza para el ADN ya que indican la ausencia de compuestos orgánicos residuales en la muestra analizada.
Tabla 3 Cuantificación y pureza de ADN extraído a partir de los diferentes métodos utilizados
| Método empleado | c (ng/µL) | Abs.260 | Abs.280 | Abs.260/280 | Abs.260/230 |
|---|---|---|---|---|---|
| QIAprep® | 48,05 | 0,961 | 0,468 | 2,05 | 2,25 |
| Precipitación etanólica | 42,24 | 0,845 | 0,454 | 1,86 | 1,50 |
| T3 (pH-5) | 38,76 | 0,775 | 0,397 | 1,95 | 0,82 |
Como se muestra en la tabla 3 la purificación mediante el soporte comercial QIAprep® Spin Miniprep Kit presenta los mejores resultados en cuanto a recobrado y calidad del ADN; sin embargo, los tres métodos permiten la obtención de un ADN plasmídico libre de contaminantes proteicos y con valores de recobrado aceptables para su uso en subsiguientes procesos biológico-moleculares. Los métodos de purificación de ADN mediante precipitación etanólica y utilizando las Fe3O4@SiO2 como soporte sólido (T3) permiten obtener un plásmido libre de contaminantes proteicos, pero con la presencia de algunos residuos orgánicos. Es posible que dichos residuos se deban a la presencia de etanol en ambos casos, la cual podría eliminarse si se incrementa el tiempo de secado luego de los lavados con etanol y antes de la re-suspensión en el buffer de elusión EB, paso previo a la separación final.
La eficacia del método de separación y aislamiento mediante Fe3O4@SiO2 obtenidas deberá ser avalado en estudios posteriores mediante la utilización del ADN purificado en aplicaciones más exigentes como la clonación, la transfección y la secuenciación.
Conclusiones
Fue posible sintetizar nanopartículas magnéticas de óxido de hierro recubiertas con sílice con tamaño inferior a los 12nm de forma sencilla y económica. El estudio mediante Rietveld arrojó la presencia de magnetita como única fase; mientras que la cantidad de Si adsorbida sobre la superficie de las partículas magnéticas fue inferior al utilizar HClO4 para la estabilización de las mismas. La compensación de pH durante la síntesis permitió obtener menores tamaños de partícula; sin embargo, este parámetro no determinó la eficiencia en la purificación del ADN. Por otro lado, se demostró que es posible recuperar ADN plasmídico libre de contaminantes proteicos utilizando como soporte sólido las Fe3O4@SiO2 sintetizadas sin compensación de pH y sin la estabilización en medio ácido

