










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La tiourea y sus derivados tienen una amplia aplicación en la medicina, la agricultura y la química analítica. Su actividad antimicrobiana, citotóxica y anti-HIV ha sido ampliamente estudiada.1
En particular, la sustitución simultánea de un átomo de hidrógeno de un grupo -NH2 del núcleo tioureido H2NC(S)NH2 por un grupo acilo o aroilo y la de uno o los dos átomos de hidrógeno del otro grupo -NH2, da lugar a la formación de las 1-(acil/aroil) tioureas 3-sustituidas, compuestos con fórmula general R1C(O)N1HC(S)N3R2R3 (figura 1), donde R puede ser un sustituyente alquilo, arilo o heterocíclico. La posibilidad de combinar tantos fragmentos moleculares de naturaleza diferente y las características estructurales propias del núcleo de estos derivados de tiourea, genera la enorme versatilidad de su aplicación, ya sea como material de partida en la síntesis orgánica de compuestos heterocíclicos o en la química de coordinación debido al elevado potencial de enlace de los grupos carbonilo y tiocarbonilo con varios iones metálicos. Por ejemplo, estos derivados de tiourea han sido empleados con fines medioambientales como ionóforos en sensores potenciométricos y amperométricos para la detección de iones Pb(II), Cd(II), Hg(II), Cu(II) y Ag(I) 2 con resultados prometedores.
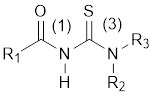
Por tanto, la construcción, caracterización y uso de electrodos de estado sólido (ESIs) basados en derivados de tiourea como receptores para la determinación de metales pesados es un tema de actualidad que involucra la selección cuidadosa del ionóforo. Entre los receptores que se han utilizado en sensores para la detección de iones Pb(II) se encuentran:1-benzoil-3,3-dimetiltiourea (1), 1-(2-furoil)-3,3-dimetiltiourea (2) y 1-(2-tiofenil)-3,3-dimetiltiourea (3) (figura 1). Aunque se ha investigado la respuesta analítica de ESIs basados en estos ionóforos 3, resulta conveniente el desarrollo de modelos teóricos que permitan esclarecer las diferencias de reactividad de estas moléculas durante el reconocimiento iónico debida a la naturaleza de los grupos sustituyentes en el núcleo de tiourea. Esta información permitiría optimizar el proceso de diseño y selección de futuros receptores al elegir los sustituyentes de este tipo de molécula mediante el proceso de síntesis. La estructura de estos compuestos ha sido ya estudiada mediante técnicas espectroscópicas y de difracción.4-6
La teoría de los funcionales de la densidad (DFT, de sus siglas en inglés) es principalmente una teoría de la estructura electrónica del estado fundamental, expresada en términos de la densidad electrónica, en lugar de la función de onda del sistema. Constituye no sólo un marco teórico en el que se desarrollan metodologías de cómputo muy eficientes, sino que permite cuantificar conceptos definidos por los orbitales moleculares frontera (OMF) tales como: potencial de ionización (I), afinidad electrónica (A), dureza global (η), potencial químico (μ), blandura (S) y electronegatividad (Χ).7 El análisis de los OMF de los derivados de tiourea, en particular la energía del HOMO, es muy importante en la comprensión de la reacción de reconocimiento del ión metálico.8 Esta interacción transcurre mediante control orbitálico donde la molécula aporta el HOMO y el ión metálico el LUMO como una típica interacción ácido-base, o electrófilo-nucléofilo. La energía del HOMO puede modularse a través de los grupos sustituyentes en el núcleo tioureido 9,10 y así influir de manera positiva en la interacción con el Pb(II).
El objetivo principal del trabajo es el estudio teórico de la estructura molecular y la absorción en el espectro infrarrojo (IR) de los compuestos (1), (2) y (3), cuyas estructuras cristalinas ya se conocen. El propósito es relacionar los parámetros moleculares calculados con los analíticos obtenidos de la caracterización de ESIs a iones Pb(II) basados en (1), (2) y (3), tales como sensibilidad, tiempo de vida y los coeficientes de selectividad potenciométricos de posibles interferentes. Para describir teóricamente los receptores se emplea el funcional B3LYP. No existen en la literatura trabajos que relacionen la respuesta analítica de los ESIs a Pb(II) con los parámetros globales derivados de los OMF para las moléculas empleadas como receptores.
Materiales y métodos
Los reactivos utilizados provienen de las firmas Sigma-Aldrich, BDH y Merck con calidad puro para análisis y sin purificación posterior. En todos los casos se trabajó con agua bidestilada (bidestilador Aquatron modelo A4D y conductividad ≤1,5 μS/cm) a temperatura ambiente de 25,0 ± 0,5 °C. Las mediciones potenciométricas se realizaron en un pHmetro digital Crisson Basic20 utilizando la siguiente composición en la celda electroquímica: Ag/AgCl| AgCl(0,1 mol/L) |KNO3 (10%) || disolución de trabajo || membrana de PVC | soporte conductor | Cu(s).
Construcción y caracterización de electrodos selectivos a iones Pb(II)
Los derivados de tiourea empleados como ionóforos o receptores (1), (2) y (3) fueron sintetizados y caracterizados por investigadores del Laboratorio de Síntesis Orgánica de la Facultad de Química, Universidad de La Habana.11 Los espectros IR de los receptores se obtuvieron en un espectrómetro FT-IR Bomem-Michelson 102, resolución de 2 cm-1 en pastillas de KBr, entre los 4000-400 cm−1. La construcción de los ESIs se realiza según la técnica reportada por Lima y Machado.12 El procedimiento seguido ha sido utilizado y reportado por Lazo-Fraga y colaboradores.13) La caracterización analítica de los electrodos selectivos a iones Pb(II) se realizó mediante la determinación de los parámetros de sensibilidad, tiempo de vida óptimo y posibles iones interferentes, entre otros, según las recomendaciones de la IUPAC.14) Este último parámetro se evaluó mediante el cálculo de los coeficientes de selectividad potenciométricos (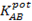 ) a partir, del Método de las Soluciones Mezcladas (MSM).15
) a partir, del Método de las Soluciones Mezcladas (MSM).15
Detalles computacionales
Las geometrías y la estructura molecular de los derivados (1), (2) y (3) en el estado basal se estudiaron por medio de cálculos que consideran todos los electrones con el funcional B3LYP 16,17 y la base estándar 6-311G(d,p). Esta última fue seleccionada porque combina flexibilidad y eficiencia computacional.18 Se utilizó el programa Gaussian 09 y el visualizador GV06 (Gaussian, Inc. USA 2016).19 Todas las estructuras optimizadas se confirmaron como mínimos locales sobre la superficie de energía potencial por presentar frecuencias positivas. Los valores de frecuencias calculados se corrigen mediante el factor de escala 0,96.20 Las asignaciones de los modos de vibración calculados se realizan con ayuda del visualizador GV06. Los cálculos se realizan con las facilidades del Shared Hierarchical Academic Research Computing Network SHARCNET and Compute/CalculCanadá.
Resultados y discusión
Las estructuras moleculares optimizadas y la numeración de los átomos se muestran en la Fig. 2. La tabla 1 reúne una selección de parámetros geométricos teóricos al nivel B3LYP/6-311G(d,p) para los compuestos (1), (2) and (3). Como referencia se proveen, además, los valores experimentales correspondientes encontrados en la literatura. En general se aprecia concordancia entre los valores reportados de las estructuras de DRX 4-6 y los obtenidos mediante los cálculos del presente estudio, validando el funcional y conjunto de base seleccionados. Esto sugiere que las geometrías de (1), (2) y (3) calculadas en fase gaseosa son similares a las que presentan en fase cristalina.
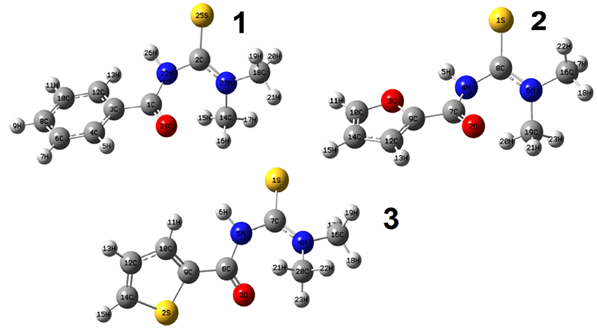
Fig. 2 Geometrías optimizadas de los receptores: 1-benzoil-3,3-dimetiltiourea (1), 1-(2-furoil)-3,3-dimetiltiourea (2) y 1-(2-tiofenil)-3,3-dimetiltiourea (3); DFT/B3LYP/6-311G(d,p).
Los enlaces C-S y C-O muestran carácter de doble enlace en todos los casos. Mientras, las distancias C-N del fragmento aciltioureido presentan valores comprendidos entre enlace simple y doble que reflejan la elevada deslocalización electrónica existente en este fragmento de las moléculas, debido al efecto de resonancia. Además, los ángulos de enlace calculados C1-N22-C2= 124,9° y C2-N23-C14=124,0° en el receptor (1), indican que los átomos de N del núcleo aciltioureido presentan hibridación sp2. De igual forma ocurre para los receptores (2) y (3).
El grado de libertad fundamental en la conformación del esqueleto principal de estos derivados está dado por la rotación alrededor del enlace (CO)N-C(S) 5,18,21, correspondiente a los enlaces C2-N22, C8-N4 y C7-N5 de los receptores (1), (2) y (3), respectivamente. Dada su mayor longitud, este enlace presenta una menor barrera rotacional por lo que estos compuestos suelen presentar un esqueleto no plano según evidencian los valores del ángulo diedro C(O)-N-C(S)-N en la Tabla 1. La típica conformación espacial "S-torcida" que adoptan los grupos carbonilo y tiocarbonilo en las 1-aciltioureas 3,3-disustituidas se presenta en la figura 2.
Tabla 1 Algunos parámetros geométricos optimizados (distancias de enlace [Å], ángulos, ángulos diedro [°] y enlaces de hidrógeno) de los receptores (1), (2) y (3); DFT/B3LYP/6-311G(d,p).
| Distancias de enlace [Å] | |||||||||||
| (1) | Calc. | Exp. [4] | (2) | Calc. | Exp. [5] | (3) | Calc. | Exp. [6] | |||
| C1-O24 | 1,214 | 1,213 | C7-O2 | 1,216 | 1,221 | C8-O3 | 1,216 | 1,221 | |||
| C1-N22 (N-H) | 1,401 | 1,390 | C7-N4 (N-H) | 1,395 | 1,387 | C8-N5 (N-H) | 1,401 | 1,374 | |||
| C2-S25 | 1,679 | 1,676 | C8-S1 | 1,678 | 1,687 | C7-S1 | 1,679 | 1,679 | |||
| C2-N22 | 1,413 | 1,396 | C8-N4 | 1,412 | 1,404 | C7-N5 | 1,413 | 1,417 | |||
| C2-N23 (-CH3) | 1,342 | 1,321 | C8-N6 (-CH3) | 1,342 | 1,321 | C7-N4 (-CH3) | 1,341 | 1,323 | |||
| Ángulos de enlace [°] | |||||||||||
| (1) | Calc. | Exp. [4] | (2) | Calc. | Exp. [5] | (3) | Calc. | Exp. [6] | |||
| O24-C1-N22 | 123,0 | 122,3 | O2-C7-N4 | 124,8 | 123,7 | O3-C8-N5 | 123,6 | 122,5 | |||
| O24-C1-C3 | 122,3 | 122,9 | O2-C7-C5 | 121,4 | 122,6 | O3-C8-C9 | 122,3 | - | |||
| C1-N22-C2 | 124,9 | 123,8 | C7-N4-C8 | 125,1 | 123,9 | C8-N5-C7 | 124,8 | 123,5 | |||
| N22-C2-N23 | 116,7 | 116,9 | N4-C8-N6 | 116,5 | 117,5 | N5-C7-N4 | 116,7 | 116,9 | |||
| N23-C2-S25 | 125,1 | 123,9 | N6-C8-S1 | 125,2 | 123,5 | N4-C7-S1 | 125,2 | 125,6 | |||
| N22-C2-S25 | 118,2 | 119,2 | N4-C8-S1 | 118,3 | 118,9 | N5-C7-S1 | 118,1 | 117,5 | |||
| C2-N23-C14 | 124,0 | 120,5 | C8-N6-C19 | 123,9 | 120,1 | C7-N4-C20 | 124,0 | - | |||
| Ángulos diedro [°] | |||||||||||
| (1) | Calc. | Exp. [4] | (3) | Calc. | Exp. [6] | ||||||
| O24-C1-N22-C2 | -12,3 | -2,60 | O3-C8-N5-C7 | 12,9 | -9,40 | ||||||
| C1-N22-C2 -N23 | 54,8 | 57,9 | C8-N5-C7 -N4 | -54,6 | 69,0 | ||||||
| C3-C1-N22 -C2 | 168 | 177 | C9-C8-N5-C7 | -167 | 171 | ||||||
| S25-C2-N22-C1 | -127 | -124 | S1-C7-N5-C8 | 127 | -113 | ||||||
| N22-C2-N23-C18 | -171 | -174 | N5-C7-N4-C20 | -17,4 | 10,2 | ||||||
| S25-C2-N23-C18 | 10,3 | 8,60 | S1-C7-N4-C20 | -10,5 | 3,10 | ||||||
| N22-C2-N23-C14 | 17,1 | 10,6 | N5-C7-N4-C16 | 171 | -179 | ||||||
| Enlaces de hidrógeno [Å] | |||||||||||
| (1) | Calc. | Exp. [4] | (2) | Calc. | Exp. [5] | (3) | Calc. | Exp. [6] | |||
| (N22-26···S25) | 2,634 | 2,650 | (N4-H5···S1) | 2,672 | 2,425 | (N5-H6···S1) | 2,630 | 2,730 | |||
| (C14-16···O24) | 2,473 | 2,380 | (C19-H21···O2) | 2,485 | 2,431 | (C20-H23···O3) | 2,474 | - | |||
Análisis vibracional
En la tabla 2 aparecen los principales valores de frecuencias experimentales para los compuestos (1), (2) y (3), las frecuencias vibracionales armónicas calculadas, así como las asignaciones tentativas de estos modos vibracionales.
Tabla 2 Principales frecuencias armónicas vibracionales calculadas -no escaladas y escaladas- B3LYP/6-311G(d,p) y su comparación con las frecuencias experimentales para los receptores (1), (2) y (3); [cm-1]; factor de escala= 0,96 20,22
| Receptor - 1 | ||||||
|---|---|---|---|---|---|---|
| νexp (cm-1) [8] | νcalc(cm-1) no escalada | Δ1 (νcalc-νexp) | νcalc(cm-1) escalada: 0,96 | Δ1(νcalc/escalada- νexp) | Asignación tentativa | |
| 3200 | 3592 | 392 | 3448 | 248 | ν(N-H) | |
| 3058 | 3184 | 126 | 3057 | -1 | ν(C-H)ar | |
| 3026 | 3152 | 126 | 3026 | 0 | νas(C-H)met | |
| 3000 | 3096 | 96 | 2972 | -28 | νas(C-H)met | |
| 2910 | 3040 | 130 | 2918 | 8 | νs(C-H)met | |
| 2864 | 3024 | 160 | 2903 | 39 | νs(C-H)met | |
| 1680 | 1760 | 80 | 1690 | 10 | ν(C=O) | |
| 1600 | 1640 | 40 | 1574 | -26 | ν(C=C)ar | |
| 1550 | 1584 | 34 | 1521 | -29 | δ(N-H) + ν(C-N) | |
| 1390 | 1416 | 26 | 1359 | -31 | δ(C-N) | |
| 1180 | 1216 | 36 | 1167 | -13 | δ(C-NH) | |
| 660 | 670 | 10 | 643 | -17 | ν(C=S) | |
| Receptor - 2 | ||||||
| νexp (cm-1) [6] | νcalc(cm-1) no escalada | Δ (νcalc-νexp) | νcalc(cm-1) escalada: 0,96 | Δ1(νcalc/escalada- νexp) | Asignación tentativa | |
| 3296 | 3584 | 288 | 3441 | 145 | ν(N-H) | |
| 3162 | 3248 | 86 | 3118 | -44 | ν(C-H)ar | |
| 3050 | 3152 | 102 | 3026 | -24 | νas(C-H)met | |
| 2923 | 3040 | 117 | 2918 | -5 | νs(C-H)met | |
| 1672 | 1762 | 90 | 1692 | 20 | ν(C=O) | |
| 1608 | 1624 | 16 | 1559 | -49 | ν(C=C)ar | |
| 1537 | 1583 | 46 | 1520 | -17 | δ(N-H) | |
| 1473 | 1480 | 7 | 1421 | -52 | δ(C-H) | |
| 1263 | 1280 | 17 | 1229 | -34 | δ(CN-H) | |
| 1178 | 1214 | 36 | 1165 | -13 | ν (C-N) | |
| 664 | 723 | 59 | 694 | 30 | ν(C=S) | |
| Receptor - 3 | ||||||
| νexp (cm-1) [20] | νcalc(cm-1) no escalada | Δ (νcalc-νexp) | νcalc(cm-1) escalada: 0,96 | Δ1(νcalc/escalada- νexp) | Asignación tentativa | |
| 3296 | 3593 | 297 | 3449 | 153 | ν(N-H) | |
| 3151 | 3240 | 89 | 3110 | -41 | ν(C-H)ar | |
| 3053 | 3096 | 43 | 2972 | -81 | νas(C-H)met | |
| 2918 | 3040 | 122 | 2918 | 0 | νs(C-H)met | |
| 2852 | 3024 | 172 | 2903 | 51 | νs(C-H)met | |
| 1678 | 1750 | 72 | 1680 | 2 | ν(C=O) | |
| 1550 | 1586 | 36 | 1523 | -27 | δ(N-H) | |
| 1469 | 1480 | 11 | 1421 | -48 | δ(C-H) | |
| 1243 | 1456 | 213 | 1398 | 155 | ν(C=C)ar | |
| 1178 | 1216 | 38 | 1167 | -11 | δ(CN-H) | |
| 657 | 700 | 43 | 672 | 15 | ν(C=S) | |
Se conoce que existe un error sistemático en el cálculo de las frecuencias armónicas vibracionales y el escalado de dichas frecuencias permite mejorar la estimación con respecto a los valores experimentales. El escalado considera la anarmonicidad del sistema, que es desestimada en el modelo del oscilador armónico empleado para el cálculo de las frecuencias. En este trabajo, los números de onda de los modos vibracionales calculados al nivel B3LYP/6-311G(d,p) se escalaron mediante la propuesta de N. Sundaraganesan et al. para corregir el error teórico (factor de escala empleado 0,96).20,22 Aunque el uso del factor de escala permitió un mejor estimado de los números de onda calculados, estos aún difieren de los valores experimentales, a pesar de que la correlación es lineal (figura 3).
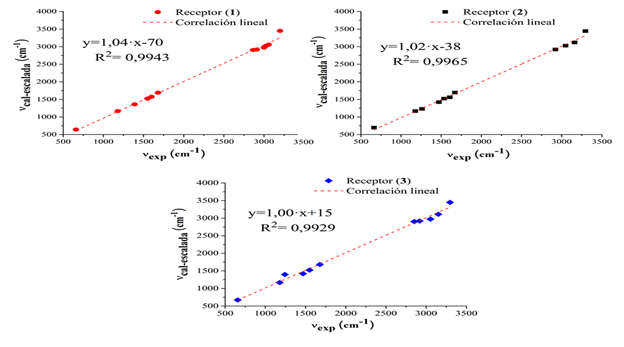
Fig. 3 Correlación lineal entre los principales números de onda calculados escalados- B3LYP/6- 311G(d,p)- y los valores observados de los espectros IR de los receptores (1), (2) y (3)
La figura 4 muestra los espectros experimentales y teóricos no escalados obtenidos para las tres moléculas.
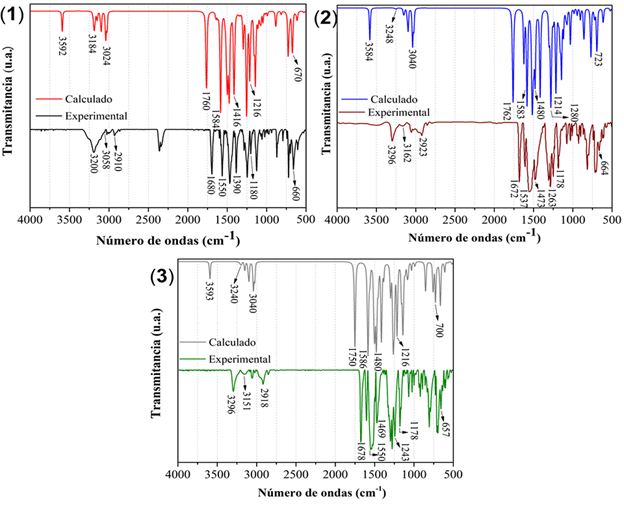
Fig. 4 Espectros IR experimentales y calculados no escalados B3LYP/6-311G(d,p) de los receptores (1), (2) y (3)
Las frecuencias vibracionales experimentales de los tres compuestos difieren fundamentalmente en aquellas bandas originadas por los modos de vibración que aportan los distintos anillos aromáticos de los grupos aroilo. Las vibraciones debidas a los estiramientos ν(N-H) aparecen de forma habitual en la región 3500-3300 cm-1.23 Las bandas de intensidad media en los espectros IR en 3200 cm-1 para (1) y en 3296 cm-1 para (2) y (3) se asocian a los modos vibracionales ν(N-H), que reflejan la influencia del heteroátomo en los anillos del grupo aroilo para (2) y (3) desplazando la posición de la banda a mayores frecuencias. Los números de onda calculados (escalados) para este modo de vibración para (1), (2) y (3) fueron 3448, 3441 y 3449 cm-1, respectivamente. Las señales en 1680, 1672 y 1678 cm-1 se asignan al modo de estiramiento ν(C=O) (calculados y escalados en 1690, 1692 y 1680 cm-1). Esta asignación coincide con lo visto en la literatura para otros derivados de tiourea relacionados.24
Las bandas debidas a los estiramientos ν(=C-H)ar en los anillos aromáticos se encuentran en el intervalo 3162-3058 cm-1; la mayor frecuencia la tiene la banda del compuesto (2), comportamiento típico de los anillos de 5 miembros (furano y tiofeno) respecto a sus análogos de 6 miembros (benceno). (25) En cuanto a las bandas débiles producto de los estiramientos simétrico (νs(C-H)met) y antisimétrico (νas(C-H)met) de los sustituyentes metilo en el núcleo tioureido, estos aparecen en el intervalo 3050-3000 cm-1 y 2923-2852 cm-1.
Es conocido que, en la tiourea y sus derivados, los enlaces C-N y C=S participan con hasta seis modos vibracionales, y que estos aparecen muchas veces acoplados.26 Por ello, en los compuestos que contienen el grupo tioamida, HN-(C=S)-N, se identifican las llamadas bandas “tioamida” I, II, III y IV que poseen las siguientes contribuciones; ν(C-N) + δ(N-H) (I), ν(C-N) + ν(C=S) (II y III) y ν(C=S) (IV). Así, las intensas bandas asociadas a los doblajes en el plano δ(CN-H) aparecen en 1550, 1537 y 1550 cm-1 para (1), (2) y (3), mientras que las debidas a los doblajes δ(C-NH) se presentan en 1180, 1263 y 1178 cm-1, respectivamente. En la región entre los 1500-1050 cm-1 se observan varias bandas debidas a doblajes en el plano δ(C-H) con fuerte acoplamiento con modos de estiramiento ν(C-C) de los anillos aromáticos.
Finalmente, la banda debida al ν(C=S), de gran importancia en este tipo de derivados por ser el azufre el principal centro de coordinación con los iones metálicos se asigna en los 660, 664 y 657 cm-1 para los compuestos (1), (2) y (3). Según los valores calculados y escalados este modo de vibración estaría en 643, 694, y 672 cm-1. La asignación de esta señal es una tarea ardua debido a su baja intensidad y al encontrarse en una región muy poblada en bandas, por lo que ha sido objeto de controversia en la literatura pues otros autores la asignan por encima de los 1000 cm-1.23 No obstante, en algunos estudios se ha observado el desplazamiento de la banda debido a la coordinación con metales de transición hacia la región 650-750 cm-1.25
Cargas atómicas
En la Tabla 3 aparecen algunas cargas atómicas calculadas por el método de orbitales naturales de enlace (NBO, de sus siglas en inglés) para los tres receptores considerados.
Tabla 3 Principales cargas atómicas NBO para los receptores (1),(2) y(3) en fase gaseosa; B3LYP/ 6-311G(d,p).
| Grupos funcionales | (1) | (2) | (3) | ||||
|---|---|---|---|---|---|---|---|
| -C=O | O24 | -0,599 | O2 | -0,596 | O3 | -0,598 | |
| C1 | 0,696 | C7 | 0,644 | C8 | 0,664 | ||
| N-H | N22 | -0,666 | N4 | -0,655 | N5 | -0,659 | |
| H26 | 0,410 | H5 | 0,419 | H6 | 0,408 | ||
| C=S | C2 | 0,300 | C8 | 0,295 | C7 | 0,299 | |
| S25 | -0,243 | S1 | -0,237 | S1 | -0,241 | ||
| N-CH3 | N23 | -0,411 | N6 | -0,412 | N4 | -0,411 | |
| C14 | -0,353 | C19 | -0,352 | C20 | -0,353 | ||
| H16 | 0,213 | H21 | 0,213 | H23 | 0,214 | ||
| O3(furoil) | -0,463 | S2(tiofenil) | 0,475 | ||||
Los valores de carga negativa, en orden decreciente de magnitud, corresponden al: N(N-H)> O(C=O)> N(>CH3)>C(CH3) > S(C=S). Mientras que los valores de carga positiva, están sobre: C(C=O)> H (N-H)> C(C=S)> H (CH3). Esta distribución de cargas sugiere la formación de enlace de hidrógeno (N-H···S y C-H···O) intra e intermolecular en fase cristalina tal como demuestran los datos referentes a enlaces de hidrógeno que aparecen en la Tabla 1, y en correspondencia con la información experimental.4-6
Orbitales moleculares frontera (OMF)
Se conoce que el orbital molecular ocupado de mayor energía (HOMO, de sus siglas en inglés) gobierna la capacidad de la molécula de donar electrones, mientras que el orbital molecular desocupado de menor energía (LUMO, de sus siglas en inglés) describe la capacidad del compuesto de aceptar electrones.26 Además, la diferencia de energía ΔEHOMO-LUMO (E gap ) se ha usado como indicador directo de la estabilidad en cuanto a ganar o perder electrones en un proceso. Un alto valor de Egap implica alta estabilidad y baja reactividad química, porque es energéticamente desfavorable adicionar electrones a un orbital LUMO alto o extraer electrones de un orbital HOMO bajo.27 Los valores de energía de los orbitales frontera EHOMO y ELUMO, así como la Egap correspondiente a cada receptor estudiado se muestran en la figura 5.
Los conceptos químicos cualitativos tales como dureza, blandura, pueden ser definidos por el E gap . El herramental de DFT ha probado ser exitoso en la determinación de tales parámetros.7 En la tabla 4 se encuentran los parámetros calculados a partir de los valores de las energías de los OMF y las expresiones empleadas para su determinación.
En todos los casos el potencial químico es negativo lo que indica que los receptores son estables, o sea, no se descomponen espontáneamente. Se aprecia que (3) presenta la menor EHOMO (en valor absoluto), con relación a los receptores (1) y (2), por lo que el orbital está más disponible para su interacción con el LUMO del ion metálico. El compuesto (3) que presenta la menor Egap y según 27 es el más reactivo y menos estable. Es también el receptor de mayor S. En cambio, los receptores (1) y (2) muestran valores similares y superiores de la E HOMO .
En la tabla 4 aparece el momento dipolo total calculado (M). Los valores obtenidos reflejan la influencia de los heteroátomos, de los anillos furánico y tiofénico, en la mayor polarización de las moléculas, siendo más significativa en el compuesto (2) por la mayor electronegatividad del átomo de oxígeno. Este descriptor puede ser importante para analizar la compatibilidad del ionóforo con el disolvente mediador. En general se conoce que una mayor lipofilicidad del receptor puede favorecer el proceso de reconocimiento por una mayor afinidad con el disolvente mediador o plastificante utilizado en la preparación de la membrana sensora.28 En este caso, la solubilidad debe favorecerse para las moléculas (1) y (3) por sus momentos dipolo menores. No obstante, las moléculas estudiadas son similares y consideramos que no es en este caso un factor determinante en la respuesta del sensor.
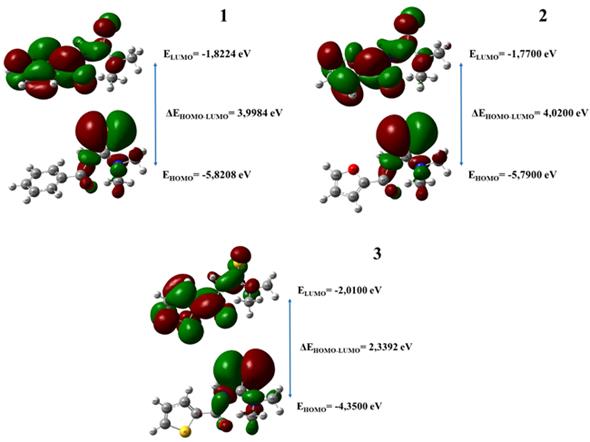
Fig. 5 Representación de los orbitales HOMO y LUMO y su gap correspondiente para los receptores (1), (2) y (3) calculados al nivel B3LYP/6-311G(d,p).
Los resultados de los descriptores de reactividad calculados sugieren el siguiente orden de reactividad para las moléculas estudiadas: (3) > (1) ≈ (2). Este orden es consistente con los resultados obtenidos en la caracterización analítica de los electrodos selectivos a iones Pb(II) que emplean a (1), (2) y (3) como ionóforos (tabla 5).
En la tabla 5 se puede apreciar que los sensores basados en los receptores (1) y (2) presentan la mejor respuesta analítica. Estos dispositivos muestran una sensibilidad cercana al valor teórico reportado por Nernst para un catión divalente (29,53 mV/dec) y tiempos de vida útil de 60 y 100 días, respectivamente. En cambio, el sensor basado en el receptor (3) no presenta una sensibilidad adecuada y su tiempo de vida es mucho más corto. Además, el análisis de los coeficientes de selectividad potenciométricos (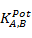 ) indica que estos son mayores en los sensores basados en el receptor (3), tanto para el ion Cd(II) como para el ion Cu(II), con valores inferiores y muy similares en el caso de los sensores basados en (1) y (2).Este hecho demuestra la menor utilidad del ESI-(3) con respecto a los otros sensores en un medio donde exista una concentración apreciable de dichos iones interferentes.
) indica que estos son mayores en los sensores basados en el receptor (3), tanto para el ion Cd(II) como para el ion Cu(II), con valores inferiores y muy similares en el caso de los sensores basados en (1) y (2).Este hecho demuestra la menor utilidad del ESI-(3) con respecto a los otros sensores en un medio donde exista una concentración apreciable de dichos iones interferentes.
Tabla 4 Descriptores de reactividad calculados para los receptores (1),(2) y (3) en fase gaseosa; DFT/B3LYP/ 6-311G(d,p).
| Descriptores | Expresión de cálculo | (1) | (2) | (3) |
| E HOMO (eV) | - | -5,820 8 | -5,790 0 | -4,350 0 |
| E LUMO(eV) | - | -1,822 4 | -1,770 0 | -2,010 0 |
| |E
|
- | 3,998 4 | 4,020 0 | 2,339 2 |
| Potencial de ionización, I(eV) |
 HOMO(1) HOMO(1) |
5,820 8 | 5,790 0 | 4,352 0 |
| Electroafinidad, A(eV) |
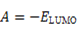 (2) (2) |
1,822 4 | 1,770 0 | 2,012 8 |
| Electronegatividad, χ (eV) |
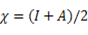 (3) (3) |
3,821 6 | 3,780 0 | 3,182 4 |
| Potencial químico, μ (eV) |
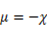 (4) (4) |
-3,821 6 | -3,780 0 | -3,182 4 |
| Dureza química, η(eV) |
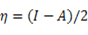 (5) (5) |
1,999 2 | 2,010 0 | 1,169 6 |
| Blandura química, S(eV-1) |
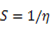 (6) (6) |
0,500 2 | 0,497 5 | 0,855 0 |
| Momento dipolo (Μ) | ||||
| Μx | 2,2694 | 2,6206 | -2,310 2 | |
| Μy | -2,1131 | -2,5082 | -1,622 6 | |
| Μz | 2,6718 | 2,1570 | 3,004 5 | |
| Momento dipolo total | 4,0932 | 4,2202 | 4,122 8 | |
Tabla 5 Parámetros analíticos determinados a electrodos de estado sólido selectivos a iones Pb(II) basados en los receptores (1), (2) y (3).
| Parámetros analíticos | (1) | (2) | (3) |
|---|---|---|---|
| Sensibilidad (mV/dec) | 30,1 ± 0,3 | 31,6 ± 0,2 | 26,5 ± 0,3 |
| Tiempo de vida útil (días) | 60 | 100 | 7 |
Coeficientes de selectividad potenciométricos (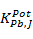 ) para las especies interferentes Cd(II) y Cu(II) ) para las especies interferentes Cd(II) y Cu(II) | |||
| Cd(II) | 1, 5 | 1,2 | 2,4 |
| Cu(II) | 3,7 | 3,4 | 4,5 |
Estos resultados justifican el orden de reactividad encontrado para los receptores estudiados. La mayor reactividad de (3) en su interacción con los iones metálicos Pb(II), Cd(II) y Cu(II) sugiere un rápido envenenamiento de la membrana sensora y afecta la reversibilidad del equilibrio en que se basa el funcionamiento de este sensor.
Conclusiones
Las geometrías de (1), (2) y (3) calculadas en fase gaseosa, son similares a las que presentan en fase cristalina y muestran la típica conformación espacial “S-torcida” que adoptan los grupos carbonilo y tiocarbonilo en las 1-aciltioureas 3,3-disustituidas. Los parámetros geométricos calculados por DFT para los tres receptores son congruentes con los obtenidos por DRX. Los números de onda de los espectros IR calculados y escalados son superiores pero comparables a los valores experimentales y su correlación es lineal. La distribución de cargas positivas y negativas (NBO) en los tres compuestos, sugieren la formación de enlaces de hidrógeno (N-H···S y C-H···O) intra e intermolecular en fase cristalina. Se encontró una correspondencia entre los parámetros de reactividad global calculados y los parámetros analíticos experimentales de los ESIs a iones Pb(II). El orden de reactividad obtenido es: (3) > (1) ≈ (2)

