










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El primer cianoacrilato de alquilo (CA) fue descubierto por el Dr. Harry Coover y sus colaboradores en 1942. Sus marcadas propiedades adhesivas permitieron su comercialización para aplicaciones industriales. Más tarde, su espectro de acción se amplió al campo de la medicina. La condensación de Knoevenagel es la reacción más empleada para su obtención, debido a su simplicidad, reproducibilidad, buenos rendimientos y alta pureza. No obstante, prosigue la investigación en su mejora, variando los distintos parámetros del proceso. La radiación de microondas (MW) como fuente de calentamiento más amigable con el medio ambiente y económica, se escogió por primera vez para la síntesis del BCA por los autores del presente trabajo, 1 al reducir los tiempos de la mayoría de las reacciones.
Las nanopartículas (NP) y las nanofibras (NF) de PCA son nuevas formas de presentación de los CA con potencialidades para aplicaciones en el campo médico. Las NP de PCA presentan una toxicidad relativamente baja, capaces de cruzar diferentes barreras biológicas, transportar distintos tipos de fármacos y relativamente fáciles de sintetizar. Desde finales de los años 70 hasta hoy,2 se investiga la introducción de dichos sistemas en la práctica social para el tratamiento de distintas dolencias. Lo más avanzado, es el fármaco Livatag®, basado en NP de policianoacrilato de hexilo cargadas con doxorrubicina para el tratamiento de carcinoma hepatocelular.3
Las NF, por su parte, fueron descubiertas más recientemente, con pocas publicaciones.4-12 La polimerización en fase vapor y el electro-hilado se han empleado para ello, sin embargo, esta última es la utilizada con propósitos médicos. El electro-hilado se ha aplicado a CA mezclados con polimetacrilato de metilo10-12 o a CA diluidos en disolventes polares8,9 para promover la polimerización in situ de CA sobre órganos, tejidos y dispositivos médicos en forma de NF. En el 2020 se publicó, por los autores del presente trabajo, una patente relacionada con la obtención de NF de PCA con el fin de utilizarlas en forma de mallas para distintas aplicaciones médicas como apósitos, transportadores de fármacos y en ingeniería de tejidos.13
A continuación, se describen los métodos utilizados para la síntesis del BCA y sus polímeros, PBCA de distintas masas moleculares, así como para la obtención de micro/nanofibras de PBCA. Además, se muestra la influencia de algunos parámetros de la técnica de electro-hilado sobre la morfología de las mallas y ejemplos de aplicabilidad en la medicina.
MÉTODOS
Reactivos
Todos los reactivos y disolventes, a excepción del cianoacetato de n-butilo, fueron usados sin purificación previa y adquiridos de firmas certificadas bajo la categoría “para síntesis”.
Síntesis del cianoacrilato de n-butilo
La condensación de Knoevenagel entre el cianoacetato de n-butilo (0,3517 mol) y el formaldehido (0,5275 mol) en presencia de tolueno (0,3000 mol) e hidrocloruro de piperidina (0,0020 mol) se realizó mediante 2 vías cuya diferencia fue la fuente de calentamiento. El baño de aceite incluyó 4 etapas: a) calentamiento de la mezcla de reacción (130 oC) con recogida de la mezcla tolueno-agua, b) extracción de los restos de la mezcla bajo presión reducida, c) pirólisis del producto con pentóxido de fósforo (0,01 mol), hidroquinona (0,004 mol) y ácido p-tolueno sulfónico (0,002 mol) y d) purificación del producto y su estabilización con hidroquinona (0,002 mol) y ácido p-tolueno sulfónico (0,0005 mol). Con el horno de microondas comercial de laboratorio (Milestone’s START System, Japón) se siguió el mismo procedimiento, eliminando la segunda y cuarta etapas. En la primera toda la mezcla tolueno-agua se extrajo (200 W, 5 min y 250 W, 15 min a T máxima de 100 oC) y en la siguiente se pirolizó (650 W, 5 min y 700 W, 10 min, T máxima 200 oC, presión reducida), obteniendo el producto con la misma apariencia y poder adhesivo que el purificado al utilizar el baño de aceite.
Síntesis de policianoacrilatos de n-butilo
El PBCA ( M n =103 Da) se obtuvo de la mezcla oligomérica formada en la primera etapa de síntesis del BCA por MW. Los PBCA en los órdenes de 104, 105 y 106 Da se sintetizaron mediante la polimerización aniónica en masa del BCA según la técnica descrita por Han y colaboradores.14 Los productos se disolvieron, precipitaron sobre agua, se filtraron, secaron y se almacenaron para la producción de las nanofibras.
Determinación de la masa molecular de los policianoacrilatos de n-butilo
Se utilizó la técnica cromatográfica por permeación de gel (GPC, Waters Alliance e2695) con detectores UV e IR (concentración polimérica es igual a 2 mg/ml, eluyente es igual a THF, estándar externo es igual a PS, temperatura del horno es igual a 25 oC, volumen de inyección igual a 100 μL y flujo del eluyente igual a1 ml/min).
Caracterización química
Se utilizaron las técnicas espectroscópicas de infrarrojo y resonancia magnética nuclear. Los espectros infrarrojos del BCA y el resto de las muestras se registraron en un espectrómetro JASCOFT/IR-4100 (Japón) y Nicolet Magna IR 550 de la firma Thermo Scientific (Alemania) respectivamente. El BCA se analizó en pastillas de KBr y el resto de las muestras con ATR-FTIR a resoluciones de 2 cm-1 y 4 cm-1 respectivamente de 400 cm-1 a 4000 cm-1. Los espectros de RMN se registraron en un espectrómetro AV400 Bruker (Alemania), con frecuencias de 400 MHz para 1H y 100 MHz para 13C. Los corrimientos químicos se expresaron en ppm respecto al tetrametilsilano en todos los casos y se utilizó acetona-d6 o CDCl3.
Síntesis y caracterización de micro/nanofibras de policianoacrilatos de n-butilo
La técnica de electro-hilado tradicional se empleó para la producción de las micro/nanofibras poliméricas utilizando un equipo de laboratorio montado por los propios autores, compuesto por una fuente de alto voltaje (0-30 kV; Grom KSE 100 (D)), un sistema de inyección de jeringa (0-600 ml/h; Infors AG (CH)) y un colector estático conectado a tierra. Para obtener nanofibras se utilizaron distintos valores de los parámetros: distancia entre la punta de la aguja y el colector (5 cm - 12 cm), voltaje (8,0 kV - 21,0 kV) y velocidad de flujo (0,01 ml/min - 1,00 ml/min). Además, se prepararon disoluciones poliméricas basadas en PBCA 100 % y mezclas de disoluciones de PBCA y del citostático 5-fluorouracilo (5FU). Los disolventes utilizados (acetona pura o mezclada con dimetilformamida o etanol) se encuentran en las clases 2 y 3 de los empleados en productos farmacéuticos. Las concentraciones poliméricas se prepararon en el intervalo de 0,5 % a 80 % según la masa molecular del PBCA así como el disolvente. La concentración másica del 5FU respecto al PBCA fue entre 5 % y 30 %. La caracterización química se realizó de igual manera que para las muestras de polímero.
La microscopia electrónica de barrido (SEM) (Maxim, Cambridge-United Kingdom) se utilizó para determinar la morfología de las mallas. La máquina de ensayo mecánico universal (Zwick/Z010, Germany) equipada con una celda de 200 N de carga se utilizó para realizar el ensayo de tracción uniaxial, a T = 25 oC, p igual a 1 atm, velocidad de 1 mm/segundo y distancia de trabajo de 10 mm. Al menos se ensayaron 5 piezas rectangulares de cada malla con dimensiones de 10 mm de ancho, 15 mm de largo y 300 µm de espesor. Este último se midió de forma aproximada mediante el microscopio óptico Infinite Focus? G4 (Alicona Imaging, Grambach/Graz, Austria).
La degradación térmica de las mallas se analizó por termogravimetría (TGA) con un analizador Q500 (TA Instruments), bajo atmósfera inerte de N2 a una velocidad de flujo de 10 ml/min en el intervalo de temperatura de 22 oC a 600 oC a una velocidad de calentamiento de 10 oC/min.
Estudio de liberación del 5-fluorouracilo
Se escogieron una malla compuesta 100 % por PBCA y otra del mismo PBCA y 15 % de 5FU. Se incubaron a 37 oC, 3 muestras (7 mg) de cada una de las mallas en disolución buffer de fosfato (PBS) de pH 7,4. Se aplicó el muestreo estático y se usó espectroscopia UV (UV-mini 1240, Shimadzu, Japan) a 280 nm para la cuantificación.
Estudio de proliferación celular
Los fibroblastos humanos gingivales (HFIB-G, Provitro AG, Alemania) se cultivaron hasta el pase 4 en su medio basal compuesto por 90 % McCoy's 5ª, 5 % de albumina de suero bobino, 2 % de L-glutamina, 2 % de buffer HEPES y 1 % de penicilina/streptomicina, todos provenientes de Biochrom GmbH (Alemania).
La viabilidad celular se realizó visualmente por 2 vías. La primera, mediante la tinción de las células con el par diacetato de fluoresceína/bromuro de etidio, distinguiendo las vivas de las muertas por su color verde y rojo respectivamente bajo el microscopio de fluorescencia Olympus (VANOX-T, Japón) a 488 nm a los 3, 7, 14 y 21 días de incubación. En la segunda vía se utilizó SEM. Después de 23 días de incubación las células se fijaron con glutaraldehido al 2,5 % para su medición.
RESULTADOS Y DISCUSIÓN
Síntesis del cianoacrilato de n-butilo
El rendimiento de 66 % y la pureza, mayor que 99 % logrados al emplear la radiación de microondas fueron similares a los alcanzados por el baño de aceite con reducción del tiempo del proceso de 3 a 5 veces como consecuencia de la disminución de los pasos de la síntesis;1 lo cual pudiera deberse a una combinación de efectos térmicos y no térmicos provocados por la radiación.15
Síntesis de policianoacrilatos de n-butilo
Los rendimientos oscilaron entre 80 % y 90 %. En la mayoría de los PBCA se comprobó la proporcionalidad inversa entre la concentración del iniciador y la M n .
Masa molecular de los policianoacrilatos de n-butilo
Los valores de M n e índice de polidispersión del PBCA producto de la condensación de Knoevenagel fueron 4,0·103 Da y 1,6 respectivamente. Los correspondientes a los PBCA obtenidos mediante la polimerización aniónica en masa fueron 1,2·104 Da (1,4); 7,4·104 Da (1,7); 2,5·105 Da (4,5) y 1,1·106 Da*; 8,0·103 Da (2,7; 2,7), donde * denota la población polimérica de mayor proporción.
Caracterización química del cianoacrilato de n-butilo y los policianoacrilatos de n-butilo
No se observaron diferencias significativas entre los espectros FTIR de los PBCA de distintas M n independientemente del método de producción, pero sí con el correspondiente al BCA. La banda en 1 747 cm-1 junto a la de 1 253 cm-1 sugiere la presencia de un grupo éster. Las vibraciones C =O, C-O y C≡N muestran un corrimiento respecto a las del monómero. Además, la intensidad de las bandas asociadas a la vibración C≡N de los PBCA son menores que la del monómero, efecto explicado por la pérdida de conjugación entre esos grupos con el doble enlace.16 Por otra parte, las bandas en 3122 cm-1 y 1610 cm-1 asociadas al enlace doble están presentes únicamente en el espectro del monómero.
Las señales de los protones de los grupos metilos de todos los PBCA se desplazaron de forma similar (0,90 ppm) a los correspondientes al monómero (0,96 ppm) y sólo para los PBCA de baja masa molecular (4,0·103 y 1,2·104 Da) los protones de los grupos metilenos de la cadena lateral alquílica mostraron desplazamientos similares (1,40 ppm; 1,70 ppm) a los correspondientes al BCA (1,44 ppm; 1,72 ppm), puesto que para los PBCA con mayor masa molecular (1,1·106; 2,5·105 y 7,4·104) estas 2 señales aparecieron superpuestas en una sola centrada en 1,50 ppm. Esto es típico cuando se emplean técnicas tradicionales de RMN para analizar polímeros de alta masa molecular con acoplamientos vecinales de protones, provocando superposiciones de multipletes. Por otro lado, no aparecen las señales correspondientes a los protones del grupo vinilo del monómero en los espectros correspondientes a los polímeros, pero sí una señal centrada en 2,50 ppm asignada a los protones del grupo metileno de la cadena principal de los PBCA, confirmando el 100 % de conversión en la polimerización.
Electro-hilado de disoluciones poliméricas basadas en policianoacrilatos de n-butilo-100 %. Efecto de la concentración del policianoacrilatos de n-butilo
Para todo el intervalo de M n de los PBCA se observó que al incrementar la concentración polimérica ocurrió una transformación morfológica de nanopartículas a nanofibras, fenómeno reportado con anterioridad para otros polímeros y relacionado con la tensión superficial y la viscosidad.17,18 Además, desaparecieron los defectos estructurales: perlas, partículas y bifurcaciones (figura 1), debido al aumento del enmarañamiento entre las cadenas poliméricas y en consecuencia la fuerza viscoelástica.( )

Electro-hilado de disoluciones poliméricas basadas en policianoacrilatos de n-butilo-100 %. Efecto del disolvente
El disolvente de las disoluciones poliméricas exhibió un efecto directo sobre la sección transversal de las fibras. Así, con acetona pura, las fibras se obtuvieron planas como cintas o cilindros aplanados, mientras que con mezclas de acetona:etanol y acetona:dimetilformamida (DMF) las fibras resultaron redondas y macizas (figura 2) debido a la velocidad de evaporación de los disolventes. Cuando la disolución polimérica se prepara en acetona pura, el interior del chorro de polímero cargado se seca con mayor rapidez, conformando un cilindro hueco depositado en forma de cinta o cilindro aplanado por efecto de la presión atmosférica.20 Sin embargo, al usar mezclas de acetona con etanol o DMF la velocidad de la evaporación del disolvente es más lenta, con reducción homogénea del diámetro del chorro y recolección de fibras redondas y macizas.21

Electro-hilado de disoluciones poliméricas basadas en policianoacrilatos de n-butilo-100 %. Efecto del voltaje aplicado
El efecto del voltaje aplicado sobre el diámetro promedio de las fibras estuvo relacionado con el tipo de disolvente utilizado (figura 3). Al emplear la acetona pura, el aumento del voltaje incrementó el diámetro de las fibras casi 2 veces. Este efecto se ha observado muy poco para otros polímeros y se ha explicado porque aumenta más el flujo del chorro y la rapidez con que las fibras se depositan,21 y el disolvente dispone de menos tiempo para evaporarse.18 Sin embargo, otra explicación se corresponde mejor con los resultados del presente trabajo. Las fibras formadas como cilindros huecos durante la trayectoria hacia el colector se abren, puesto que al aumentar el voltaje aumenta la repulsión entre las cargas eléctricas, así como también la velocidad de evaporación de la acetona, de acuerdo con lo reportado por Ghorani.22 En el caso de emplear mezclas acetona:etanol, el aumento del voltaje disminuyó el diámetro de las fibras, efecto observado con mayor frecuencia,17 porque aumenta la repulsión entre las cargas con un mayor estiramiento del chorro polimérico cargado.23
Electro-hilado de disoluciones poliméricas basadas en mezclas de policianoacrilatos de n-butilo y 5-fluorouracilo. Efecto de la concentración del 5-fluorouracilo
Se obtuvieron mallas libres de imperfecciones para cada una de las mezclas PBCA:5FU utilizadas, sin marcadas diferencias en cuanto a la morfología de las fibras, independientemente de la relación másica polímero:citostático.
La malla compuesta por PBCA y 5FU al 15 % (PBCA:15 %-5FU) se caracterizó químicamente mediante FTIR y RMN-13C. El espectro infrarrojo de la malla presentó una única señal diferente respecto al PBCA, ubicada a 1 675 cm-1 debido al grupo C = O del 5FU. En su espectro RMN-13C aparecen las señales propias de sus componentes a los mismos desplazamientos químicos que en sus espectros independientes. Por otro lado, se determinaron las propiedades tensiles y térmicas de esta malla y la compuesta 100 % por PBCA (PBCA-100 %). El módulo de Young de la malla PBCA-100 % fue 1,2 ± 0,4 MPa, mientras que para PBCA:15 %-5FU resultó ligeramente inferior 0,7 ± 0,4 MPa. Ello indica que el 5FU en la malla PBCA-100 % no afecta significativamente su resistencia a la deformación elástica. Por otro lado, la tensión máxima de la malla PBCA:15 %-5 FU fue casi el doble (0,10 ± 0,03 MPa) que el de la malla PBCA-100 % (0,05 ± 0,02 MPa), indicando que la malla PBCA:15 %-5FU exhibe una mayor capacidad para soportar tensiones. Este efecto pudiera relacionarse con la distorsión que causan las moléculas de 5FU en la arquitectura de la malla PBCA-100 %, provocando una mayor porosidad (60 % vs. 39 %).

Fig. 3 Efecto del voltaje sobre el diámetro promedio (Φf) y la morfología de las fibras compuestas por PBCA-1,1·106 Da en correspondencia con el disolvente utilizado: A) mezcla de Acetona: Etanol y B) Acetona pura. Barra =2 μm en todas las fotos SEM.
Las curvas termogravimétricas de esas 2 mallas y del 5FU mostraron un decrecimiento continuo con el incremento de la temperatura. La derivada de la pérdida de peso respecto a la temperatura de la malla PBCA-100 % mostró un solo pico, indicativo de la pureza de la malla y su degradación tomó lugar entre 110 oC y 241 oC, con un máximo a 186 oC. La degradación de la malla PBCA:15 %-5FU comenzó a 100 oC y terminó a 309 oC. Su curva derivada presentó 3 picos, el más intenso a 203 oC, asociado con el proceso de descomposición del PBCA con una pérdida de peso de 85 %, que coincide con la relación másica utilizada entre el PBCA y el 5FU para producir la malla. Los otros 2 picos a 236 oC y 285 oC se asocian a procesos de descomposición del 5FU con 5 % y 10 % de pérdida respectivamente. El termograma del 5FU mostró un 94 % de pérdida de peso a 291 oC identificado como su punto de fusión a 282 oC. Estos desplazamientos pueden deberse a la distorsión que provoca la presencia de 5FU en el PBCA y viceversa.
De acuerdo a la temperatura inicial de descomposición, la malla PBCA:15 %-5FU es más inestable que la de PBCA-100 %. Esto sugiere una reducción del empaquetamiento molecular por la presencia del 5FU respecto a la malla de PBCA-100 %, limitando las interacciones entre las cadenas y por lo tanto provocando una disminución en la temperatura inicial de descomposición. Este resultado está en concordancia con el de la tensión máxima.
Estudio de liberación del 5-fluorouracilo
En la figura 4 se muestran los perfiles de liberación de las muestras tomadas en 3 posiciones diferentes de la malla PBCA:15 %-5FU. Las pequeñas barras del error indican la homogeneidad en la composición de esta. Se pueden distinguir 3 etapas de liberación: una inicial en los primeros 5 días y 7 días hasta un 20 %, atribuible al 5FU depositado en o cerca de la superficie de las fibras; una segunda, casi lineal, más lenta entre los 8 días y 34 días, indicando un atrapamiento más efectivo del 5FU en el centro de las fibras (estas 2 etapas de liberación apuntan a una distribución “core-shell” del 5FU) y una tercera etapa, donde la velocidad de liberación del 5FU se redujo grandemente, correspondiendo al 5FU remanente.
Se probaron los modelos de Ritger-Peppas, Peppas-Sahlin y los correspondientes a las cinéticas de primero y segundo orden para determinar los parámetros cinéticos de la liberación del 5FU. Los valores medios del 5FU liberado (Mt) o las fracciones (Mt/M∞) de las 3 muestras respecto al tiempo, se graficaron de acuerdo al modelo. M∞ se consideró igual al peso teórico de 5FU (inicialmente cargado en la muestra (M0)).
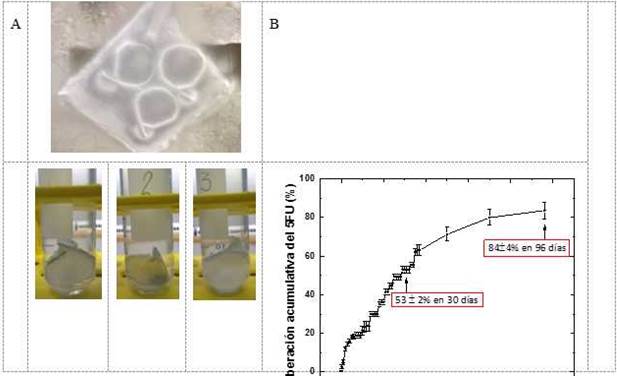
Fig. 4 A) Muestras seleccionadas de la malla PBCA:15 %-5FU para realizar el estudio de liberación del 5FU. B) perfiles de liberación del 5FU en buffer de fosfato (pH 7,4) a 37 oC hasta los 96 días de estudio.
La data experimental correspondiente a la primera etapa se ajustó al modelo empírico de Ritger-Peppas (ecuación I). La constante de velocidad de liberación (k) resultó (6,3 ± 0,5)10-2 día-1 y el exponente (n), 0,64 ± 0,05, indicando un sistema de liberación no-Fickeano. Por tanto, el mecanismo de liberación del 5FU está gobernado por una mezcla de distintos procesos.

Por otro lado, al aplicar el modelo de Peppas-Sahlin en la primera etapa (ecuación II),24 fijando las fronteras (a ≥ 0) para k 1 y k 2 , el modelo de Peppas-Sahlin se redujo al de Ritger-Peppas con k 2 = (6 ± 4)·10-2 día-1 y el exponente de difusión 0,32 ± 0,09.

La segunda etapa de liberación (de 8 días a 34 días) se ajustó a 2 modelos (Ritger-Peppas y 1er orden). El valor del exponente (n= 0,80 ± 0,03) del modelo de Ritger-Peppas corresponde a un mecanismo de liberación no-Fickeano. La constante de velocidad, (3,4 ± 0,3)10-2 día-1, resultó muy similar a la obtenida cuando se aplicó primer orden (Ecuación III) (2,53 ± 0,08)10-2 día-1, valor con orden inferior al reportado para las NP de PBCA.25 Así, el mecanismo de liberación del 5FU de la malla PBCA:15 %-5FU en la segunda etapa está gobernada por una velocidad de difusión constante.26

La tercera y última etapa de liberación se ajustó a un segundo orden respecto al 5FU remanente (ecuación IV), indicativo de un proceso de liberación gobernado por la relajación de las cadenas.27

Estudio de proliferación celular
La figura 5 muestra la forma en la que los HFIB-G se adhirieron y proliferaron sobre la malla de PBCA-100 % durante los días de incubación. Estos preservaron su forma redonda hasta el día 7, mientras que a los 14 días se observaron más fibroblastos sobre pequeñas áreas de la malla con forma alargada. En el día 21, la superficie de la malla estaba saturada de fibroblastos alineados. El 100 % de confluencia se observó en cada uno de los días de estudio en las referencias. Las mallas incubadas con y sin células después de lavadas y secadas mostraron similar apariencia, manteniendo su estructura física hasta el día 21. Las fibras se distribuyeron de forma aleatoria con muy pocos cambios (asociación y fusión de algunas fibras) y una disminución gradual de los diámetros promedio hasta el día 14 con un aumento ligero en el día 21.
Los HFIB-G incubados en las mallas de PBCA-100 % hasta los 23 días crecieron dentro del área delimitada por los tubos de acero utilizados para almacenarlas. La apariencia de la malla en diferentes posiciones fue similar tanto para las incubadas con células como en ausencia de estas.
Conclusiones
La técnica de síntesis del BCA utilizando la radiación de microondas es más económica, sencilla y segura respecto a las técnicas tradicionales de calentamiento. Es posible electro-hilar disoluciones del PBCA en un amplio intervalo de masas moleculares y producir micro/nanofibras con características diferentes de acuerdo a los valores de los parámetros que se fijen. Las mallas de micro/nanofibras de PBCA promueven la proliferación de células humanas y sirven como transportadores de fármacos para su liberación controlada.
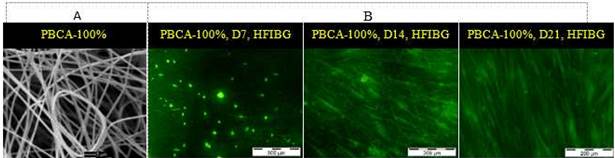
Fig. 5 A) Foto SEM de la malla PBCA-100 % seleccionada para el ensayo celular (diámetro promedio de las fibras ≈ 1 μm), Barra = 5 μm. B) Fotos de fluorescencia del crecimiento de HFIB-G sobre la malla PBCA-100 % durante el tiempo de incubación, día 7 (D7), día 14 (D14) y día 21 (D21). Barra = 500 μm (día 7) y 200 μm (días 14 y 21).

